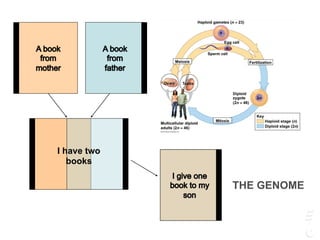
The document discusses a webinar focusing on the genomic potential of domestic cats, highlighting candidate regions under selection and the importance of single nucleotide polymorphisms (SNPs) for disease and phenotype association discovery. It emphasizes the need for a comprehensive SNP map to facilitate research into hereditary diseases and traits across various cat breeds. The findings aim to enhance understanding of genetic determinants and assist in developing genotyping platforms for feline health management.








![Mullikin et al. BMC Genomics 2010, 11:406
http://www.biomedcentral.com/1471-2164/11/406
Open AccessDATABASE
Database
Light whole genome sequence for SNP discovery
across domestic cat breeds
James C Mullikin*1, Nancy F Hansen1, Lei Shen2, Heather Ebling2, William F Donahue2, Wei Tao2, David J Saranga2,
Adrianne Brand2, Marc J Rubenfield2, Alice C Young1, Pedro Cruz1 for NISC Comparative Sequencing Program1,
Carlos Driscoll3, Victor David3, Samer WK Al-Murrani4, Mary F Locniskar4, Mitchell S Abrahamsen4, Stephen J O'Brien3,
Douglas R Smith2 and Jeffrey A Brockman4
Abstract
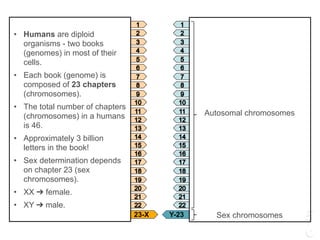
Background: The domestic cat has offered enormous genomic potential in the veterinary description of over 250
hereditary disease models as well as the occurrence of several deadly feline viruses (feline leukemia virus -- FeLV, feline
coronavirus -- FECV, feline immunodeficiency virus - FIV) that are homologues to human scourges (cancer, SARS, and
AIDS respectively). However, to realize this bio-medical potential, a high density single nucleotide polymorphism (SNP)
map is required in order to accomplish disease and phenotype association discovery.
Description: To remedy this, we generated 3,178,297 paired fosmid-end Sanger sequence reads from seven cats, and
combined these data with the publicly available 2X cat whole genome sequence. All sequence reads were assembled
together to form a 3X whole genome assembly allowing the discovery of over three million SNPs. To reduce potential
false positive SNPs due to the low coverage assembly, a low upper-limit was placed on sequence coverage and a high
lower-limit on the quality of the discrepant bases at a potential variant site. In all domestic cats of different breeds:
female Abyssinian, female American shorthair, male Cornish Rex, female European Burmese, female Persian, female
Siamese, a male Ragdoll and a female African wildcat were sequenced lightly. We report a total of 964 k common SNPs
suitable for a domestic cat SNP genotyping array and an additional 900 k SNPs detected between African wildcat and
domestic cats breeds. An empirical sampling of 94 discovered SNPs were tested in the sequenced cats resulting in a
SNP validation rate of 99%.
Conclusions: These data provide a large collection of mapped feline SNPs across the cat genome that will allow for the
development of SNP genotyping platforms for mapping feline diseases.
Background
Along with dogs, the domestic cat enjoys extensive veter-
inary surveillance, more than any other animal. A rich lit-
erature of feline veterinary models reveals a unique
opportunity to explore genetic determinants responsible
ion to people since their original domestication from the
Asian wildcat (Felis silvestris lybica), recently estimated at
approximately 10,000 years ago in the Middle East's Fer-
tile Crescent[3]. In spite of our affection for cats,
advances in clinical resolution of genetic maladies and
10.1101/gr.6380007Access the most recent version at doi:
2007 17: 1675-1689Genome Res.
Bourque, Glenn Tesler, NISC Comparative Sequencing Program and Stephen J. O’Brien
Antunes, Marilyn Menotti-Raymond, Naoya Yuhki, Jill Pecon-Slattery, Warren E. Johnson, Guillaume
A. Schäffer, Richa Agarwala, Kristina Narfström, William J. Murphy, Urs Giger, Alfred L. Roca, Agostinho
Sante Gnerre, Michele Clamp, Jean Chang, Robert Stephens, Beena Neelam, Natalia Volfovsky, Alejandro
Joan U. Pontius, James C. Mullikin, Douglas R. Smith, Agencourt Sequencing Team, Kerstin Lindblad-Toh,
Initial sequence and comparative analysis of the cat genome
data
Supplementary
http://www.genome.org/cgi/content/full/17/11/1675/DC1
"Supplemental Research Data"
References
http://www.genome.org/cgi/content/full/17/11/1675#References
This article cites 97 articles, 41 of which can be accessed free at:
service
Email alerting
click heretop right corner of the article or
Receive free email alerts when new articles cite this article - sign up in the box at the
Notes
http://www.genome.org/subscriptions/
go to:Genome ResearchTo subscribe to
© 2007 Cold Spring Harbor Laboratory Press
Comparative analysis of the domestic cat genome
reveals genetic signatures underlying feline
biology and domestication
Michael J. Montaguea,1
, Gang Lib,1
, Barbara Gandolfic
, Razib Khand
, Bronwen L. Akene
, Steven M. J. Searlee
,
Patrick Minxa
, LaDeana W. Hilliera
, Daniel C. Koboldta
, Brian W. Davisb
, Carlos A. Driscollf
, Christina S. Barrf
,
Kevin Blackistonef
, Javier Quilezg
, Belen Lorente-Galdosg
, Tomas Marques-Bonetg,h
, Can Alkani
, Gregg W. C. Thomasj
,
Matthew W. Hahnj
, Marilyn Menotti-Raymondk
, Stephen J. O’Brienl,m
, Richard K. Wilsona
, Leslie A. Lyonsc,2
,
William J. Murphyb,2
, and Wesley C. Warrena,2
a
The Genome Institute, Washington University School of Medicine, St. Louis, MO 63108; b
Department of Veterinary Integrative Biosciences, College of
Veterinary Medicine, Texas A&M University, College Station, TX 77843; c
Department of Veterinary Medicine & Surgery, College of Veterinary Medicine,
University of Missouri, Columbia, MO 65201; d
Population Health & Reproduction, School of Veterinary Medicine, University of California, Davis, CA 95616;
e
Wellcome Trust Sanger Institute, Hinxton CB10 1SA, United Kingdom; f
National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health,
Bethesda, MD 20886; g
Catalan Institution for Research and Advanced Studies, Institute of Evolutionary Biology, Pompeu Fabra University, 08003
Barcelona, Spain; h
Centro de Analisis Genomico 08028, Barcelona, Spain; i
Department of Computer Engineering, Bilkent University, Ankara 06800, Turkey;
j
Department of Biology, Indiana University, Bloomington, IN 47405; k
Laboratory of Genomic Diversity, Center for Cancer Research, Frederick, MD 21702;
l
Dobzhansky Center for Genome Bioinformatics, St. Petersburg State University, St. Petersburg 199178, Russia; and m
Oceanographic Center, Nova
Southeastern University, Fort Lauderdale, FL 33314
Edited by James E. Womack, Texas A&M University, College Station, TX, and approved October 3, 2014 (received for review June 2, 2014)
Little is known about the genetic changes that distinguish
domestic cat populations from their wild progenitors. Here we
describe a high-quality domestic cat reference genome assembly
and comparative inferences made with other cat breeds, wildcats,
and other mammals. Based upon these comparisons, we identified
positively selected genes enriched for genes involved in lipid
Previous studies have assessed breed differentiation (6, 7),
phylogenetic origins of the domestic cat (8), and the extent of
recent introgression between domestic cats and wildcats (9, 10).
However, little is known regarding the impact of the domesti-
cation process within the genomes of modern cats and how this
compares with genetic changes accompanying selection identified in
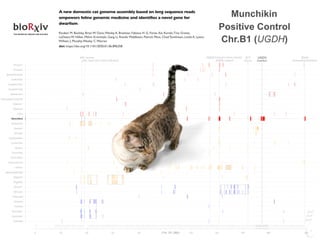
GENETICS](https://image.slidesharecdn.com/hasanwinn2020-201207114157/85/Unique-pages-in-the-cat-s-book-10-320.jpg)

























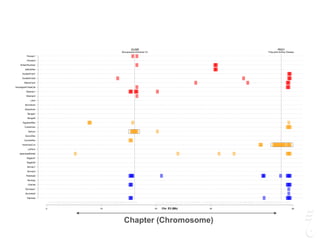
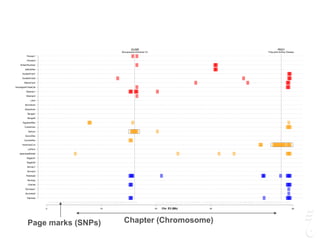
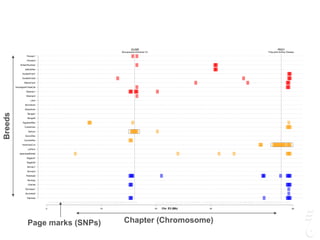
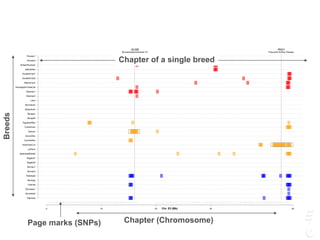
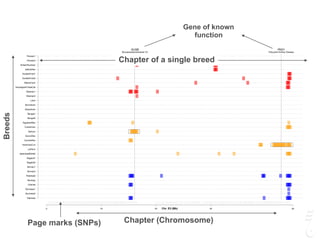
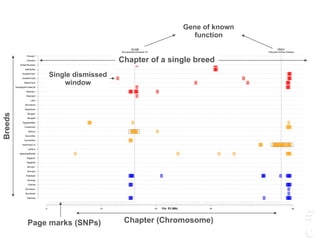
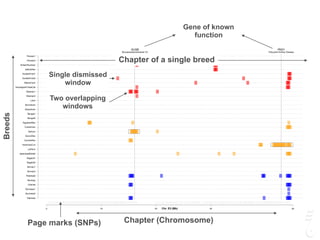
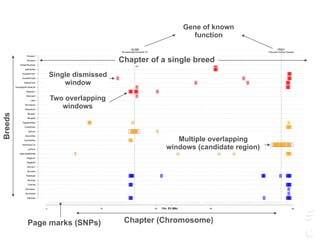


![LPAR6 Rexing
HEXB Gangliosidosis 2
ARSB Mucopolysaccharidosis VI
FXII Factor XII DeficiencyATP7B Copper Metabolism LVRN Tabby
0 20 40 60 80 100 140 160 180 200 220 243
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
Chr. A1 (Mb)
To the Root of the Curl: A Signature of a Recent Selective
Sweep Identifies a Mutation That Defines the Cornish
Rex Cat Breed
Barbara Gandolfi1
*, Hasan Alhaddad1
, Verena K. Affolter2
, Jeffrey Brockman3
, Jens Haggstrom4
,
Shannon E. K. Joslin1
, Amanda L. Koehne2
, James C. Mullikin5
, Catherine A. Outerbridge6
,
Wesley C. Warren7
, Leslie A. Lyons1
1 Department of Population Health and Reproduction, School of Veterinary Medicine, University of California - Davis, Davis, California, United States of America,
2 Department of Pathology, Microbiology, Immunology, School of Veterinary Medicine, University of California - Davis, Davis, California, United States of America, 3 Hill’s
Pet Nutrition Center, Topeka, Kansas, United States of America, 4 Department of Clinical Sciences, Faculty of Veterinary Medicine and Animal Science, Swedish University
of Agricultural Sciences, Uppsala, Sweden, 5 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research Institute, National Institutes of
Health, Bethesda, Maryland, United States of America, 6 Department of Veterinary Medicine & Epidemiology, School of Veterinary Medicine, University of California - Davis,
Davis, California, United States of America, 7 The Genome Institute, Washington University School of Medicine, St. Louis, Missouri, United States of America
Abstract
The cat (Felis silvestris catus) shows significant variation in pelage, morphological, and behavioral phenotypes amongst its
over 40 domesticated breeds. The majority of the breed specific phenotypic presentations originated through artificial
selection, especially on desired novel phenotypic characteristics that arose only a few hundred years ago. Variations in coat
texture and color of hair often delineate breeds amongst domestic animals. Although the genetic basis of several feline coat
colors and hair lengths are characterized, less is known about the genes influencing variation in coat growth and texture,
especially rexoid – curly coated types. Cornish Rex is a cat breed defined by a fixed recessive curly coat trait. Genome-wide
analyses for selection (di, Tajima’s D and nucleotide diversity) were performed in the Cornish Rex breed and in 11
phenotypically diverse breeds and two random bred populations. Approximately 63K SNPs were used in the analysis that
aimed to localize the locus controlling the rexoid hair texture. A region with a strong signature of recent selective sweep
was identified in the Cornish Rex breed on chromosome A1, as well as a consensus block of homozygosity that spans
approximately 3 Mb. Inspection of the region for candidate genes led to the identification of the lysophosphatidic acid
receptor 6 (LPAR6). A 4 bp deletion in exon 5, c.250_253_delTTTG, which induces a premature stop codon in the receptor,
was identified via Sanger sequencing. The mutation is fixed in Cornish Rex, absent in all straight haired cats analyzed, and is
also segregating in the German Rex breed. LPAR6 encodes a G protein-coupled receptor essential for maintaining the
structural integrity of the hair shaft; and has mutations resulting in a wooly hair phenotype in humans.
Citation: Gandolfi B, Alhaddad H, Affolter VK, Brockman J, Haggstrom J, et al. (2013) To the Root of the Curl: A Signature of a Recent Selective Sweep Identifies a
Mutation That Defines the Cornish Rex Cat Breed. PLoS ONE 8(6): e67105. doi:10.1371/journal.pone.0067105
Editor: Arnar Palsson, University of Iceland, Iceland
Received March 26, 2013; Accepted May 14, 2013; Published June 27, 2013
Copyright: ß 2013 Gandolfi et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits
unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This project was supported by the National Center for Research Resources and the Office of Research Infrastructure Programs of the National Institute
of Health through Grant Number R24 RR016094, the Winn Feline Foundation (W10-14, W11-041), the Center for Companion Animal Health at University of
California Davis (2010-09-F) (http://www.vetmed.ucdavis.edu/ccah/index.cfm), and the George and Phyllis Miller Feline Health Fund of the San Francisco
Foundation (2008-36-F). Support for the development of the Illumina Infinium Feline 63K iSelect DNA array was provided by the Morris Animal Foundation (http://
www.morrisanimalfoundation.org) via a donation from Hill’s Pet Food, Inc. The funders had no role in study design, data collection and analysis, decision to
publish, or preparation of the manuscript.
Competing Interests: JB works for a private company (Hill’s Pet Food, Inc) that partially sponsored the development of the 63k feline SNP array. The funder had
no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
* E-mail: bgandolfi@ucdavis.edu
Introduction
Phenotypic traits under strong artificial selection within cat
breeds vary from body types, muzzle shape, tail length to
aesthetically pleasant traits, such as hair color, length and texture.
Hair represents one of the defining characteristic of mammals.
Hair provides body temperature regulation, protection from
environmental elements, and adaptive advantages of camouflage,
as well as often having aesthetic value to humans. The hair follicle
has a highly complex structure with eight distinct cell layers, in
which hundreds of gene products play a key role in the hair cycle
maintenance [1,2]. In the past decade, numerous genes expressed
in the hair follicle have been identified and mutations in some of
these genes have been shown to underlie hereditary hair diseases
in humans and other mammals [3]. Hereditary hair diseases in
mammals show diverse hair phenotypes, such as sparse or short
hairs (hypotrichosis), excessive or elongated hairs (hypertrichosis),
and hair shaft anomalies, creating rexoid/woolly hairs [3–12].
Causative genes for the diseases encode various proteins with
different functions, such as structural proteins, transcription
factors, and signaling molecules. Mutations within structural
proteins, such as epithelial and hair keratins, are often associated
with hair disease. To date, mutations in several hair keratin genes
underlined two hereditary hair disorders: monilethrix, character-
PLOS ONE | www.plosone.org 1 June 2013 | Volume 8 | Issue 6 | e67105
Cornish Rex
Positive Control
Chr.A1 (LPAR6)
LPAR6 Rexing
HEXB Gangliosidosis 2
ARSB Mucopolysaccharidosis VI
FXII Factor XII DeficiencyATP7B Copper Metabolism LVRN Tabby
0 20 40 60 80 100 140 160 180 200 220 243
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
Chr. A1 (Mb)
LPAR6 Rexing
HEXB Gangliosidosis 2
ARSB Mucopolysaccharidosis VI
FXII Factor XII DeficiencyATP7B Copper Metabolism LVRN Tabby
0 20 40 60 80 100 140 160 180 200 220 243
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
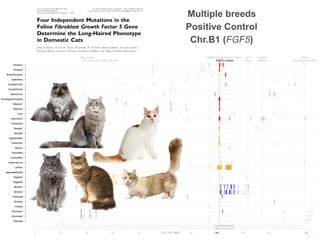
Chr. A1 (Mb)](https://image.slidesharecdn.com/hasanwinn2020-201207114157/85/Unique-pages-in-the-cat-s-book-72-320.jpg)
![FGF5 Longhair
KIT
Gloves
PKD2 Polycystic kidney disease UGDH
Dwarfism
HR Hairless IDUA
MucopolysaccharidosisLPL Lipoprotein Lipase Deficiency
0 20 40 60 80 120 140 160 180 209
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
Chr. B1 (Mb)
FGF5 Longhair
KIT
Gloves
PKD2 Polycystic kidney disease UGDH
Dwarfism
HR Hairless IDUA
MucopolysaccharidosisLPL Lipoprotein Lipase Deficiency
0 20 40 60 80 120 140 160 180 209
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
Chr. B1 (Mb)
FGF5 Longhair
KIT
Gloves
PKD2 Polycystic kidney disease UGDH
Dwarfism
HR Hairless IDUA
MucopolysaccharidosisLPL Lipoprotein Lipase Deficiency
0 20 40 60 80 120 140 160 180 209
Siamese
Burmese2
Burmese1
Oriental
Bombay
Peterbald
Birman2
Birman1
Ragdoll2
Ragdoll1
JapaneseBobtail
LaPerm
AmericanCurl
CornishRex
DevonRex
Sphynx
TurkishVan
EgyptianMau
Bengal2
Bengal1
Abyssinian
Munchkins
Lykoi
Siberian2
Siberian1
NorwegianForestCat
MaineCoon
ScottishFold2
ScottishFold1
SelkirkRex
BritishShorthair
Persian2
Persian1
Chr. B1 (Mb)
genesG C A T
T A C G
G C A T
Article
Werewolf, There Wolf: Variants in Hairless Associated
with Hypotrichia and Roaning in the Lykoi Cat Breed
Reuben M. Buckley 1,†, Barbara Gandolfi 1,†, Erica K. Creighton 1, Connor A. Pyne 1,
Delia M. Bouhan 1, Michelle L. LeRoy 1,2, David A. Senter 1,2, Johnny R. Gobble 3,
Marie Abitbol 4,5 , Leslie A. Lyons 1,* and 99 Lives Consortium ‡
1 Department of Veterinary Medicine and Surgery, College of Veterinary Medicine, University of Missouri,
Columbia, MO 65211, USA; buckleyrm@missouri.edu (R.M.B.); Barbara-Gandolfi@idexx.com (B.G.);
erica-creighton@idexx.com (E.K.C.); cap998@mail.missouri.edu (C.A.P.);
deliabouhan10@gmail.com (D.M.B.); leroymi@missouri.edu (M.L.L.); senterd@missouri.edu (D.A.S.)
2 Veterinary Allergy and Dermatology Clinic, LLC., Overland Park, KS 66210, USA
3 Tellico Bay Animal Hospital, Vonore, TN 37885, USA; jrgobblevet@gmail.com
4 NeuroMyoGène Institute, CNRS UMR 5310, INSERM U1217, Faculty of Medicine, Rockefeller,
Claude Bernard Lyon I University, 69008 Lyon, France; marie.abitbol@vetagro-sup.fr
5 VetAgro Sup, University of Lyon, Marcy-l’Etoile, 69280 Lyon, France
* Correspondence: lyonsla@missouri.edu; Tel.: +1-573-884-2287
† These authors contributed equally to this work.
‡ Membership of the 99 Lives Consortium is provided in the Acknowledgments.
Received: 12 May 2020; Accepted: 12 June 2020; Published: 22 June 2020
Abstract: A variety of cat breeds have been developed via novelty selection on aesthetic,
dermatological traits, such as coat colors and fur types. A recently developed breed, the lykoi
(a.k.a. werewolf cat), was bred from cats with a sparse hair coat with roaning, implying full color and
all white hairs. The lykoi phenotype is a form of hypotrichia, presenting as a significant reduction
in the average numbers of follicles per hair follicle group as compared to domestic shorthair cats,
a mild to severe perifollicular to mural lymphocytic infiltration in 77% of observed hair follicle groups,
and the follicles are often miniaturized, dilated, and dysplastic. Whole genome sequencing was
conducted on a single lykoi cat that was a cross between two independently ascertained lineages.
Comparison to the 99 Lives dataset of 194 non-lykoi cats suggested two variants in the cat homolog
for Hairless (HR) (HR lysine demethylase and nuclear receptor corepressor) as candidate causal gene
variants. The lykoi cat was a compound heterozygote for two loss of function variants in HR,
an exon 3 c.1255_1256dupGT (chrB1:36040783), which should produce a stop codon at amino acid
420 (p.Gln420Serfs*100) and, an exon 18 c.3389insGACA (chrB1:36051555), which should produce
a stop codon at amino acid position 1130 (p.Ser1130Argfs*29). Ascertainment of 14 additional cats
from founder lineages from Canada, France and di↵erent areas of the USA identified four additional
loss of function HR variants likely causing the highly similar phenotypic hair coat across the diverse
cats. The novel variants in HR for cat hypotrichia can now be established between minor di↵erences
in the phenotypic presentations.
Keywords: atrichia; domestic cat; Felis catus; fur; HR; naked
1. Introduction
Domestic cats have been developed into distinctive breeds during the past approximately 150 years,
since the first cat shows were held in the late 1800’s [1–3]. Many breeds have proven to be genetically
distinct [4,5] but also su↵er from inbreeding and founder e↵ects, inadvertently becoming important
biomedical models for human diseases. Over 72 diseases/traits caused by at least 115 mutations
Genes 2020, 11, 682; doi:10.3390/genes11060682 www.mdpi.com/journal/genes
Lykoi
Positive Control
Chr.B1 (HR)](https://image.slidesharecdn.com/hasanwinn2020-201207114157/85/Unique-pages-in-the-cat-s-book-73-320.jpg)

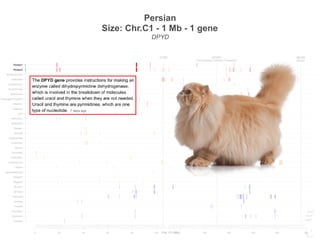




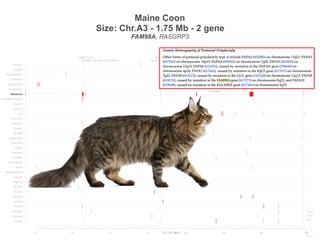

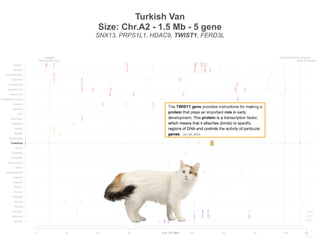
![Turkish Van
Size: Chr.A2 - 1.5 Mb - 5 gene
SNX13, PRPS1L1, HDAC9, TWIST1, FERD3L
| development in three consecutive waves, forming four dorsal hair
types: guard, awl, auchene and zigzag.[3]
The first wave starts at em-
| |
DOI: 10.1111/exd.14090
Murine dorsal hair type is genetically determined by
polymorphisms in candidate genes that influence BMP and
1
| 1
| 1
| Betoul Baz1
|
2
| 1
| Kiarash Khosrotehrani1
1
Experimental Dermatology Group, UQ
Diamantina Institute, The University of
Queensland, Brisbane, QLD, Australia
2
QIMRBerghofer Institute of Medical
Research, Brisbane, QLD, Australia
Kiarash Khosrotehrani, Experimental
Dermatology, The University of Queensland
Diamantina Institute, Translational Research
Institute, 37 Kent Street, Woolloongabba,
QLD 4102 Australia.
Email: k.khosrotehrani@uq.edu.au
Funding information
National Health and Medical Research
Council, Grant/Award Number: NHMRC. GJ.
Walker, K Khosrotehrani. Systems analysis of
skin biology and cancer, 2014-2017
Mouse dorsal coat hair types, guard, awl, auchene and zigzag, develop in three con-
secutive waves. To date, it is unclear if these hair types are determined genetically
through expression of specific factors or can change based on their mesenchymal
environment. We undertook a novel approach to this question by studying individual
hair type in 67 Collaborative Cross (CC) mouse lines and found significant variation
in the proportion of each type between strains. Variation in the proportion of zigzag,
awl and auchene, but not guard hair, was largely due to germline genetic variation.
We utilised this variation to map a quantitative trait locus (QTL) on chromosome 12
that appears to influence a decision point switch controlling the propensity for either
second (awl and auchene) or third wave (zigzag) hairs to develop. This locus contains
two strong candidates, Sostdc1 and Twist1, each of which carry several ENCODE
regulatory variants, specific to the causal allele, that can influence gene expression,
are expressed in the developing hair follicle, and have been previously reported to be
involved in regulating human and murine hair behaviour, but not hair subtype deter-
mination. Both of these genes are likely to play a part in hair type determination via
regulation of BMP and/or WNT signalling.
hair, mouse, papilla, QTL, zigzag
| Experimental Dermatology. 2020;29:450–461.wileyonlinelibrary.com/journal/exd
|
In both humans and mice there are multiple distinct hair types, each
of which differs in size and shape. Hair curl is generated largely by
kinks in the hair follicle (HF) through which the hair grows.[1]
In mice,
pelage HF development involves reciprocal mesenchymal-epithe-
lial interactions[2]
and takes place during foetal and perinatal skin
development in three consecutive waves, forming four dorsal hair
types: guard, awl, auchene and zigzag.[3]
The first wave starts at em-
bryonic day 14.5 (E14.5) and forms primary (guard) hairs. The sec-
ond wave initiated at E16.5 forms secondary (awl and auchene) hairs.
The final wave starts at E18.5 forming tertiary (zigzag) hairs.[4]
Guard
hairs have distinctively long shafts and have a sensory function.[5]
About 3% of pelage hairs are guard, ~16% awl, ~8% auchene and
polymorphisms in candidate genes that influence BMP and
1
| 1
| 1
| Betoul Baz1
|
2
| 1
| Kiarash Khosrotehrani1
© 2020 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd
1
Experimental Dermatology Group, UQ
Diamantina Institute, The University of
Queensland, Brisbane, QLD, Australia
2
QIMRBerghofer Institute of Medical
Research, Brisbane, QLD, Australia
Kiarash Khosrotehrani, Experimental
Dermatology, The University of Queensland
Diamantina Institute, Translational Research
Institute, 37 Kent Street, Woolloongabba,
QLD 4102 Australia.
Email: k.khosrotehrani@uq.edu.au
Funding information
National Health and Medical Research
Council, Grant/Award Number: NHMRC. GJ.
Walker, K Khosrotehrani. Systems analysis of
skin biology and cancer, 2014-2017
Mouse dorsal coat hair types, guard, awl, auchene and zigzag, develop in three con-
secutive waves. To date, it is unclear if these hair types are determined genetically
through expression of specific factors or can change based on their mesenchymal
environment. We undertook a novel approach to this question by studying individual
hair type in 67 Collaborative Cross (CC) mouse lines and found significant variation
in the proportion of each type between strains. Variation in the proportion of zigzag,
awl and auchene, but not guard hair, was largely due to germline genetic variation.
We utilised this variation to map a quantitative trait locus (QTL) on chromosome 12
that appears to influence a decision point switch controlling the propensity for either
second (awl and auchene) or third wave (zigzag) hairs to develop. This locus contains
two strong candidates, Sostdc1 and Twist1, each of which carry several ENCODE
regulatory variants, specific to the causal allele, that can influence gene expression,
are expressed in the developing hair follicle, and have been previously reported to be
involved in regulating human and murine hair behaviour, but not hair subtype deter-
mination. Both of these genes are likely to play a part in hair type determination via
regulation of BMP and/or WNT signalling.
hair, mouse, papilla, QTL, zigzag
Graeme J. Walker and Kiarash Khosrotehrani equally contributed as senior authors.](https://image.slidesharecdn.com/hasanwinn2020-201207114157/85/Unique-pages-in-the-cat-s-book-87-320.jpg)


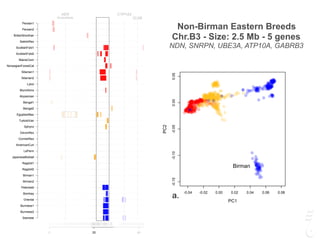



![Conference Report
Angelman Syndrome 2005: Updated Consensus for
Diagnostic Criteria
Charles A. Williams,1,2
* Arthur L. Beaudet,2,3
Jill Clayton-Smith,4
Joan H. Knoll,5
Martin Kyllerman,6
Laura A. Laan,7
R. Ellen Magenis,8
Ann Moncla,9
Albert A. Schinzel,10
Jane A. Summers,11
and Joseph Wagstaff2,12
1
Department of Pediatrics, Division of Genetics, R.C. Philips Unit, University of Florida, Gainesville, Florida
2
Scientific Advisory Committee, Angelman Syndrome Foundation, Aurora, Illinois
3
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas
4
Academic Department of Medical Genetics, St. Mary’s Hospital, Manchester, United Kingdom
5
Section of Medical Genetics and Molecular Medicine, Children’s Mercy Hospital and Clinics,
University of Missouri-Kansas City School of Medicine, Kansas City, Missouri
6
Department of Neuropediatrics, The Queen Silvia Children’s Hospital, University of Goteborg, Goteborg, Sweden
7
Department of Neurology, Leiden University Medical Center, RC Leiden, The Netherlands
8
Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, Oregon
9
De´partement de Ge´ne´tique Me´dicale, Hoˆpital des enfants de la Timone, Marseille, France
10
Institute of Medical Genetics, University of Zurich, Zurich, Switzerland
11
McMaster Children’s Hospital, Hamilton Health Sciences, Hamilton, Ontario, Canada
12
Department of Pediatrics, Clinical Genetics Program, Carolinas Medical Center, Charlotte, North Carolina
Received 19 September 2005; Accepted 2 October 2005
In 1995, a consensus statement was published for the
purpose of summarizing the salient clinical features of
Angelman syndrome (AS) to assist the clinician in making a
timely and accurate diagnosis. Considering the scientific
advances made in the last 10 years, it is necessary now
to review the validity of the original consensus criteria. As in
the original consensus project, the methodology used for
this review was to convene a group of scientists and
clinicians, with experience in AS, to develop a concise
consensus statement, supported by scientific publications
where appropriate. It is hoped that this revised consensus
document will facilitate further clinical study of individuals
with proven AS, and assist in the evaluation of those who
appear to have clinical features of AS but have normal
laboratory diagnostic testing. ß 2006 Wiley-Liss, Inc.
Key words: angelman syndrome; imprinting center;
15q11.2-q13; paternal UPD; diagnosis; criteria; behavioral
phenotype; EEG
INTRODUCTION
In 1995, a consensus statement was published for
the purpose of summarizing the salient clinical
features of Angelman syndrome (AS) [Williams
et al., 1995]. Now, a decade later, it seems appro-
priate to review these criteria in light of our increased
knowledge about the molecular and clinical features
of the syndrome. Like the first study, the methodol-
ogy used to update the revision was to convene
a group of scientists and clinicians, with experience
in AS, to develop a concise consensus statement,
supported by the scientific publications on AS. The
Scientific Advisory Committee of the U.S. AS Foun-
dation assisted in the selection of individuals who
were invited to contribute to this project.
As in the original consensus study, Tables I–III are
used here and are intended to assist in the evaluation
and diagnosis of AS, especially for those unfamiliar
with this clinical disorder. These criteria are applic-
ableforthefourknowngeneticmechanismsthatlead
to AS: molecular deletions involving the 15q11.2-q13
critical region (deletion positive), paternal unipar-
ental disomy (UPD), imprinting defects (IDs), and
mutations in the ubiquitin-protein ligase E3A gene
(UBE3A).
Table I lists the developmental history and
laboratory findings expected for AS. There are only
minor changes when compared to the original 1995
*Correspondence to: Charles A. Williams, M.D., Department of
Pediatrics, Division of Genetics, P.O. Box 100296, Gainesville, FL
32610. E-mail: Willicx@peds.ulf.edu
DOI 10.1002/ajmg.a.31074
ß 2006 Wiley-Liss, Inc. American Journal of Medical Genetics 140A:413–418 (2006)
Conference Report
Angelman Syndrome 2005: Updated Consensus for
Diagnostic Criteria
Charles A. Williams,1,2
* Arthur L. Beaudet,2,3
Jill Clayton-Smith,4
Joan H. Knoll,5
Martin Kyllerman,6
Laura A. Laan,7
R. Ellen Magenis,8
Ann Moncla,9
Albert A. Schinzel,10
Jane A. Summers,11
and Joseph Wagstaff2,12
1
Department of Pediatrics, Division of Genetics, R.C. Philips Unit, University of Florida, Gainesville, Florida
2
Scientific Advisory Committee, Angelman Syndrome Foundation, Aurora, Illinois
3
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas
4
Academic Department of Medical Genetics, St. Mary’s Hospital, Manchester, United Kingdom
5
Section of Medical Genetics and Molecular Medicine, Children’s Mercy Hospital and Clinics,
University of Missouri-Kansas City School of Medicine, Kansas City, Missouri
6
Department of Neuropediatrics, The Queen Silvia Children’s Hospital, University of Goteborg, Goteborg, Sweden
7
Department of Neurology, Leiden University Medical Center, RC Leiden, The Netherlands
8
Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, Oregon
9
De´partement de Ge´ne´tique Me´dicale, Hoˆpital des enfants de la Timone, Marseille, France
10
Institute of Medical Genetics, University of Zurich, Zurich, Switzerland
11
McMaster Children’s Hospital, Hamilton Health Sciences, Hamilton, Ontario, Canada
12
Department of Pediatrics, Clinical Genetics Program, Carolinas Medical Center, Charlotte, North Carolina
Received 19 September 2005; Accepted 2 October 2005
In 1995, a consensus statement was published for the
purpose of summarizing the salient clinical features of
Angelman syndrome (AS) to assist the clinician in making a
timely and accurate diagnosis. Considering the scientific
advances made in the last 10 years, it is necessary now
to review the validity of the original consensus criteria. As in
the original consensus project, the methodology used for
this review was to convene a group of scientists and
clinicians, with experience in AS, to develop a concise
consensus statement, supported by scientific publications
where appropriate. It is hoped that this revised consensus
document will facilitate further clinical study of individuals
with proven AS, and assist in the evaluation of those who
appear to have clinical features of AS but have normal
laboratory diagnostic testing. ß 2006 Wiley-Liss, Inc.
Key words: angelman syndrome; imprinting center;
15q11.2-q13; paternal UPD; diagnosis; criteria; behavioral
phenotype; EEG
INTRODUCTION
In 1995, a consensus statement was published for
the purpose of summarizing the salient clinical
features of Angelman syndrome (AS) [Williams
et al., 1995]. Now, a decade later, it seems appro-
priate to review these criteria in light of our increased
knowledge about the molecular and clinical features
of the syndrome. Like the first study, the methodol-
ogy used to update the revision was to convene
a group of scientists and clinicians, with experience
in AS, to develop a concise consensus statement,
supported by the scientific publications on AS. The
Scientific Advisory Committee of the U.S. AS Foun-
dation assisted in the selection of individuals who
were invited to contribute to this project.
As in the original consensus study, Tables I–III are
used here and are intended to assist in the evaluation
and diagnosis of AS, especially for those unfamiliar
with this clinical disorder. These criteria are applic-
ableforthefourknowngeneticmechanismsthatlead
to AS: molecular deletions involving the 15q11.2-q13
critical region (deletion positive), paternal unipar-
ental disomy (UPD), imprinting defects (IDs), and
mutations in the ubiquitin-protein ligase E3A gene
(UBE3A).
Table I lists the developmental history and
laboratory findings expected for AS. There are only
minor changes when compared to the original 1995
*Correspondence to: Charles A. Williams, M.D., Department of
Pediatrics, Division of Genetics, P.O. Box 100296, Gainesville, FL
32610. E-mail: Willicx@peds.ulf.edu
DOI 10.1002/ajmg.a.31074
UPD [Robinson et al., 1996, 2000]. While it does
appear that fetal development and prenatal studies
such as ultrasound and growth parameters remain
normal in AS, it has recently been discovered that
assisted reproductive technologies (ART), such as
gue thrusting, and poor breast attachment. In later
infancy, gastroesophageal reflux can occur. Such
feeding abnormalities can also occur in other
neurological disorders so its presence is quite non-
specific regarding raising increased suspicion for the
TABLE II. 2005: Clinical Features of AS
A. Consistent (100%)
. Developmental delay, functionally severe
. Movement or balance disorder, usually ataxia of gait, and/or tremulous movement of limbs. Movement disorder can be mild. May not
appear as frank ataxia but can be forward lurching, unsteadiness, clumsiness, or quick, jerky motions
. Behavioral uniqueness: any combination of frequent laughter/smiling; apparent happy demeanor; easily excitable personality, often with
uplifted hand-flapping, or waving movements; hypermotoric behavior
. Speech impairment, none or minimal use of words; receptive and non-verbal communication skills higher than verbal ones
B. Frequent (more than 80%)
. Delayed, disproportionate growth in head circumference, usually resulting in microcephaly (2 SD of normal OFC) by age 2 years.
Microcephaly is more pronounced in those with 15q11.2-q13 deletions
. Seizures, onset usually 3 years of age. Seizure severity usually decreases with age but the seizure disorder lasts throughout adulthood
. Abnormal EEG, with a characteristic pattern, as mentioned in the text. The EEG abnormalities can occur in the first 2 years of life and can
precede clinical features, and are often not correlated to clinical seizure events
C. Associated (20%–80%)
. Flat occiput
. Occipital groove
. Protruding tongue
. Tongue thrusting; suck/swallowing disorders
. Feeding problems and/or truncal hypotonia during infancy
. Prognathia
. Wide mouth, wide-spaced teeth
. Frequent drooling
. Excessive chewing/mouthing behaviors
. Strabismus
. Hypopigmented skin, light hair, and eye color compared to family), seen only in deletion cases
. Hyperactive lower extremity deep tendon reflexes
. Uplifted, flexed arm position especially during ambulation
. Wide-based gait with pronated or valgus-positioned ankles
. Increased sensitivity to heat
. Abnormal sleep-wake cycles and diminished need for sleep
. Attraction to/fascination with water; fascination with crinkly items such as certain papers and plastics
. Abnormal food related behaviors
. Obesity (in the older child)
. Scoliosis
. Constipation](https://image.slidesharecdn.com/hasanwinn2020-201207114157/85/Unique-pages-in-the-cat-s-book-99-320.jpg)






















































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)








