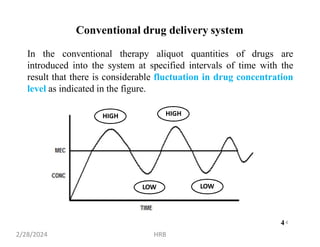
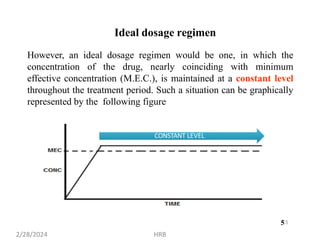
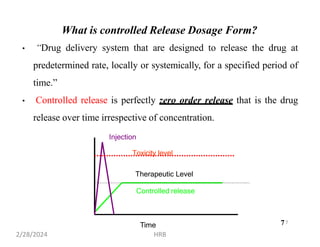
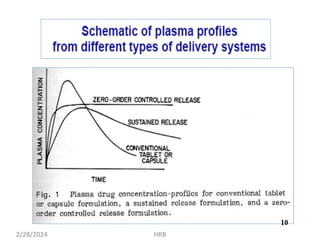
This document provides information about sustained and controlled drug delivery systems. It begins with definitions of sustained release and controlled release, and discusses the advantages of maintaining consistent drug levels over time. It then covers topics like steady state concepts, diffusion mechanisms, dissolution models and polymer characterization as they relate to sustained and controlled release drug delivery. Evaluation methods for sustained release and controlled release tablets are also mentioned.












































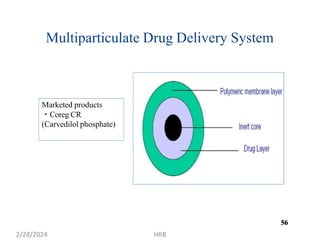

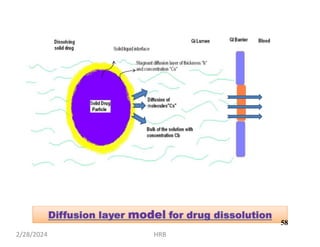


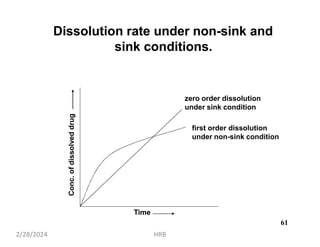




















































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)






