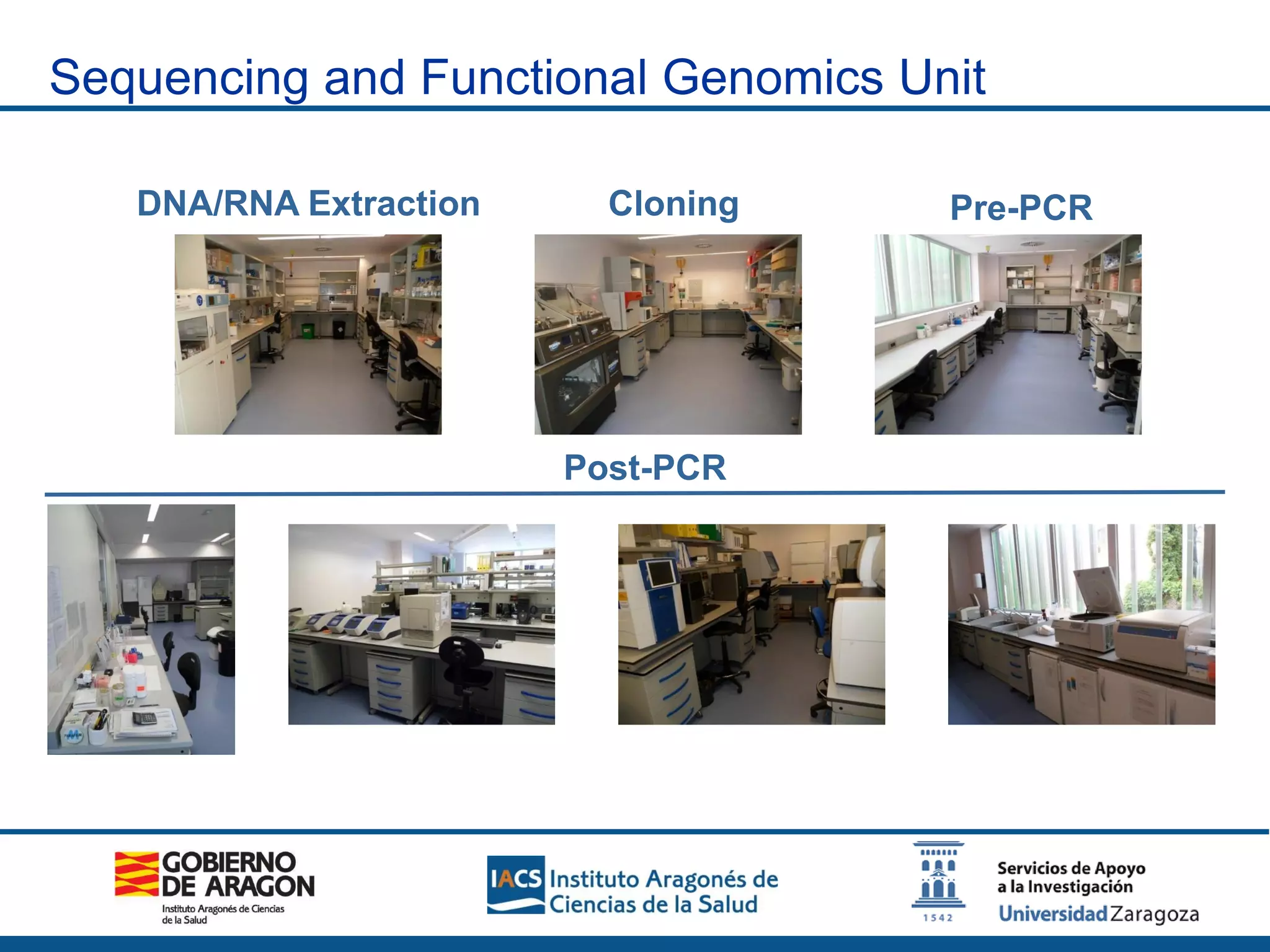
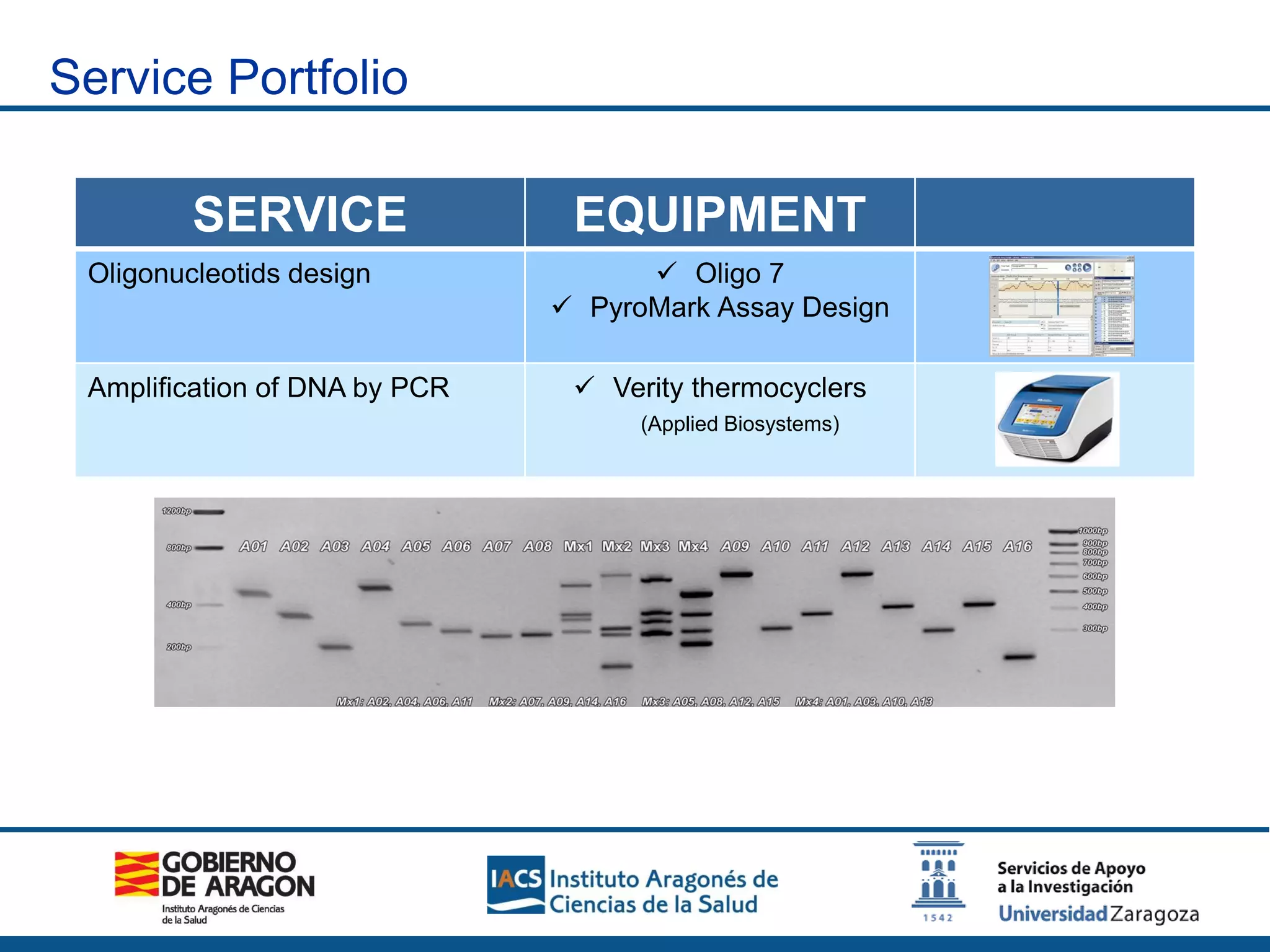
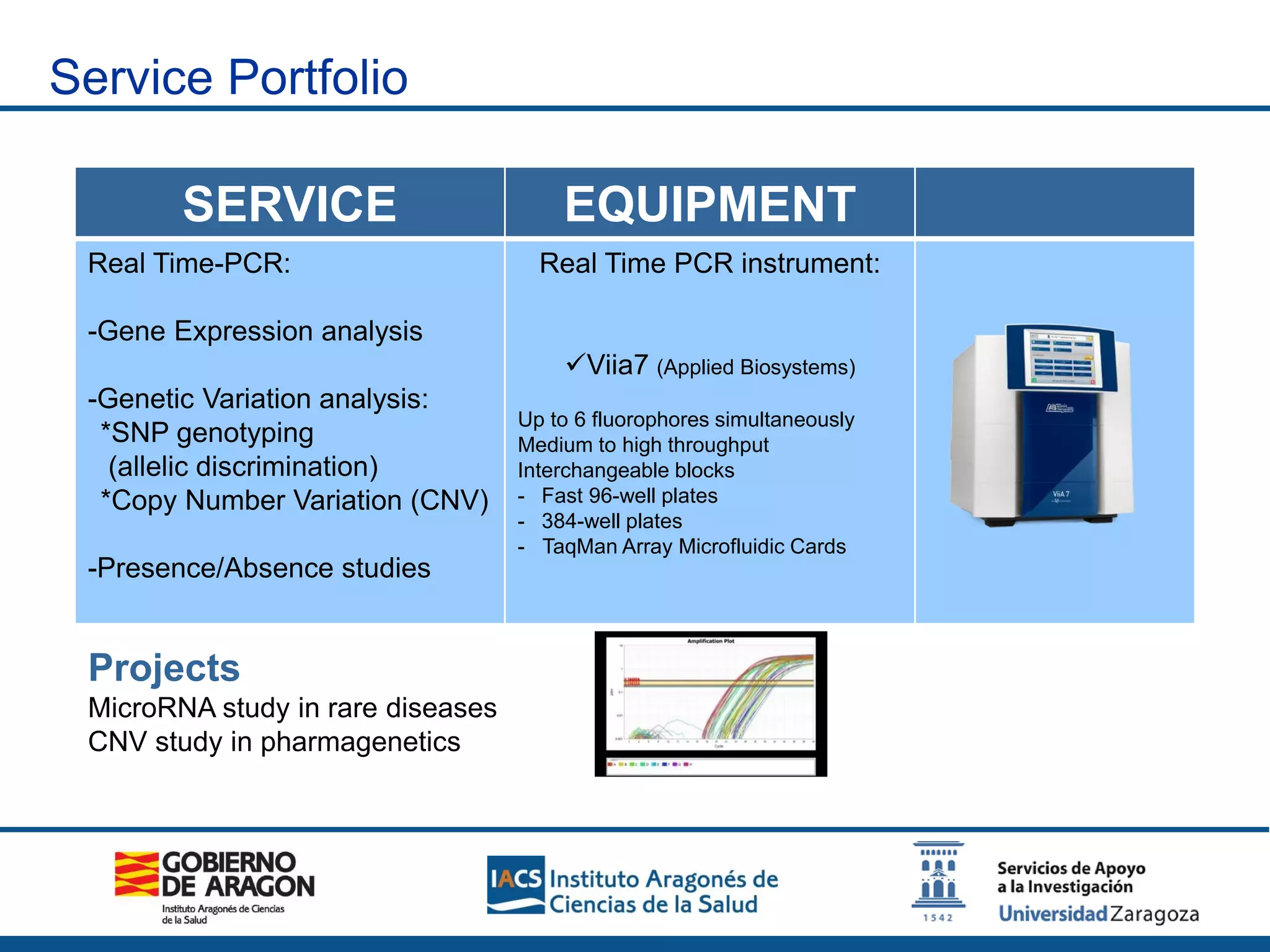
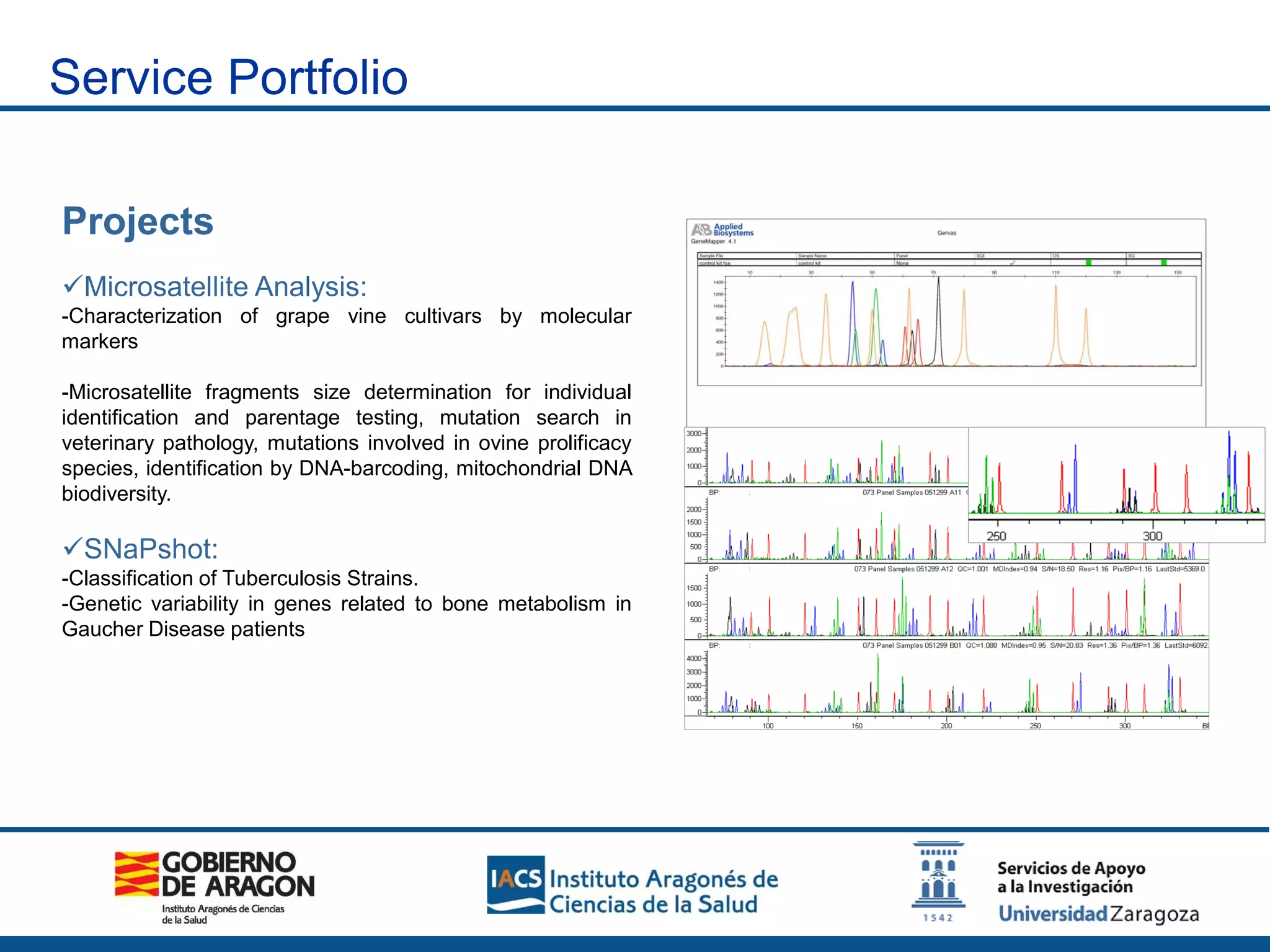
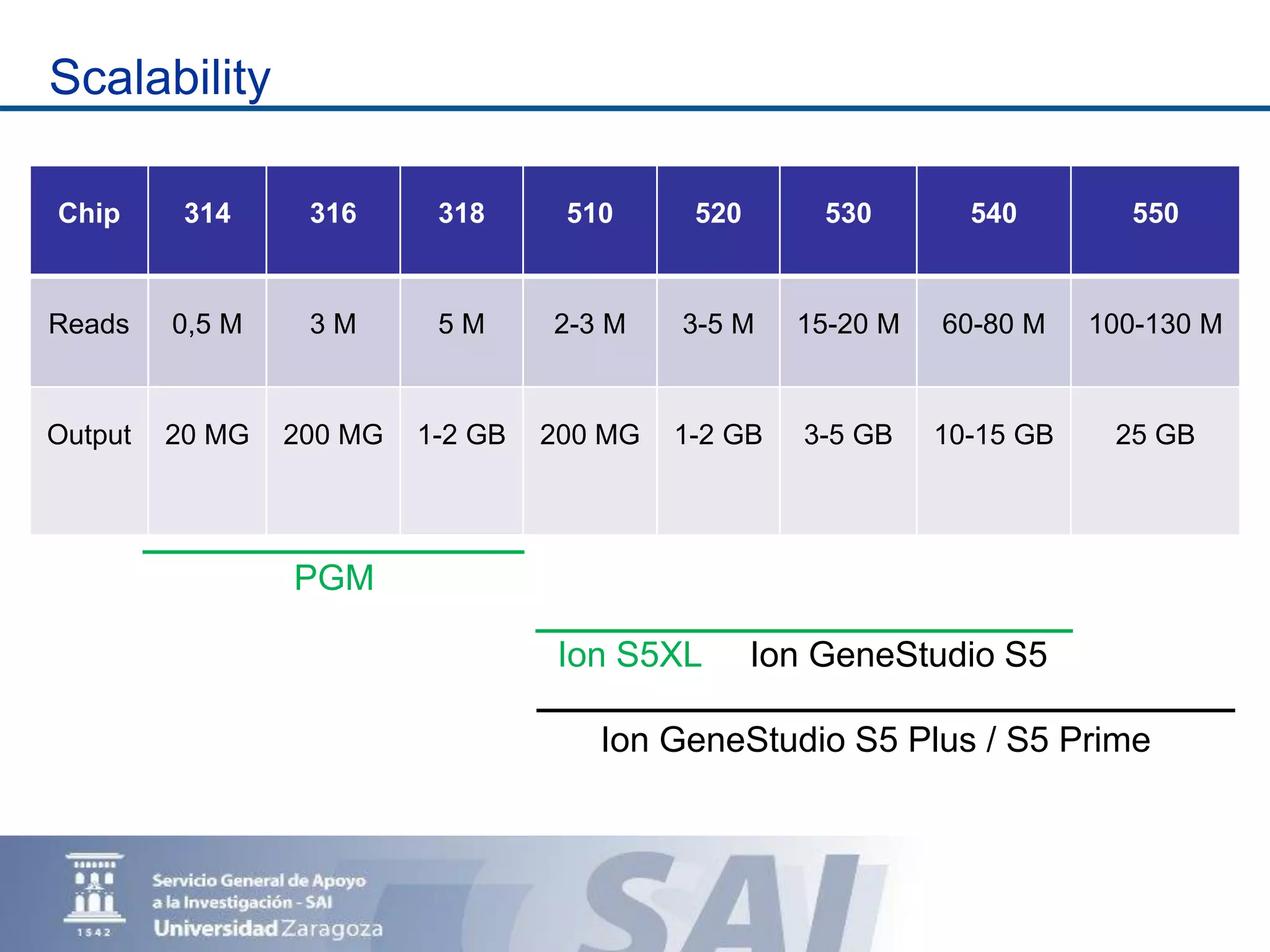
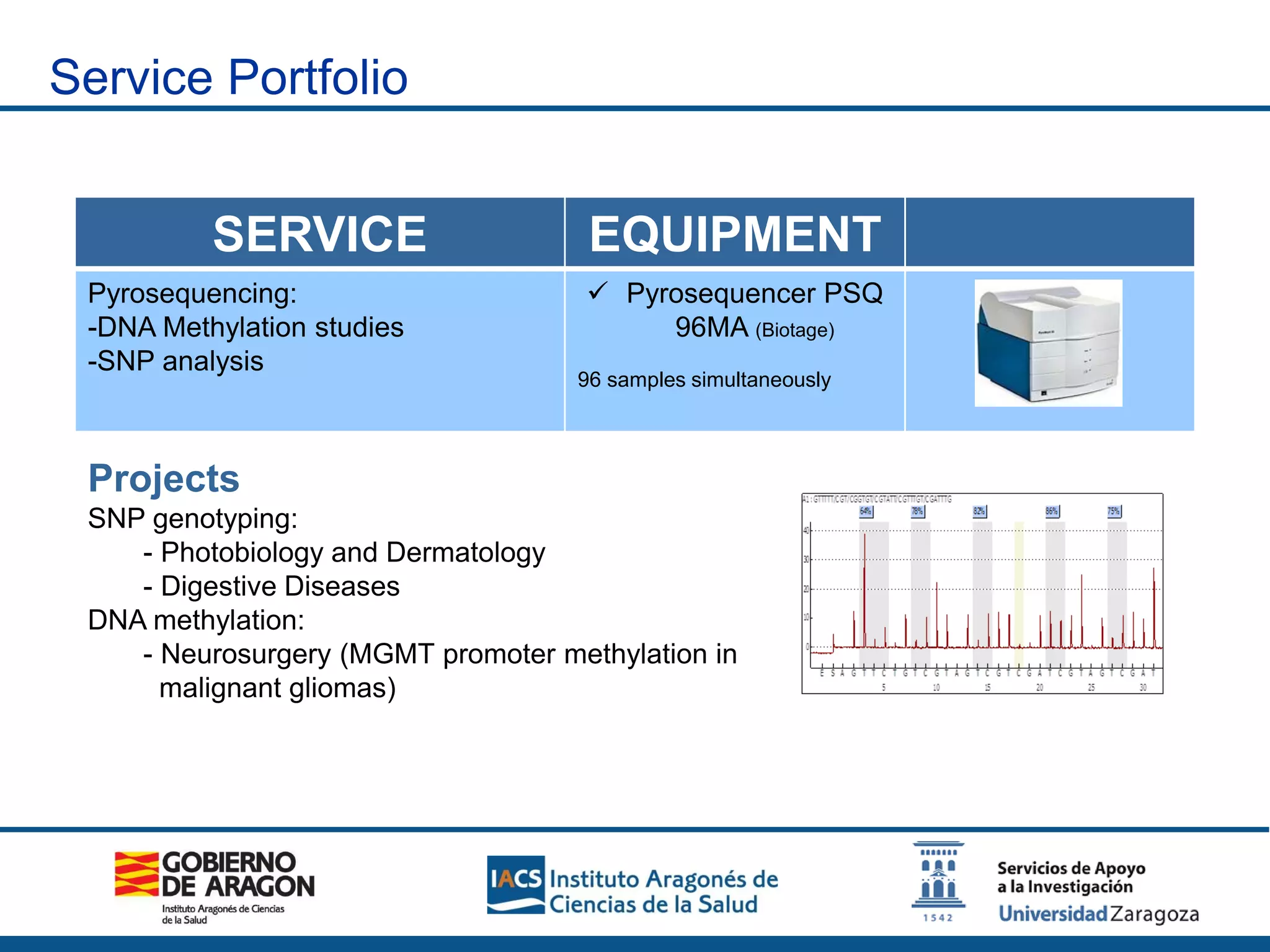
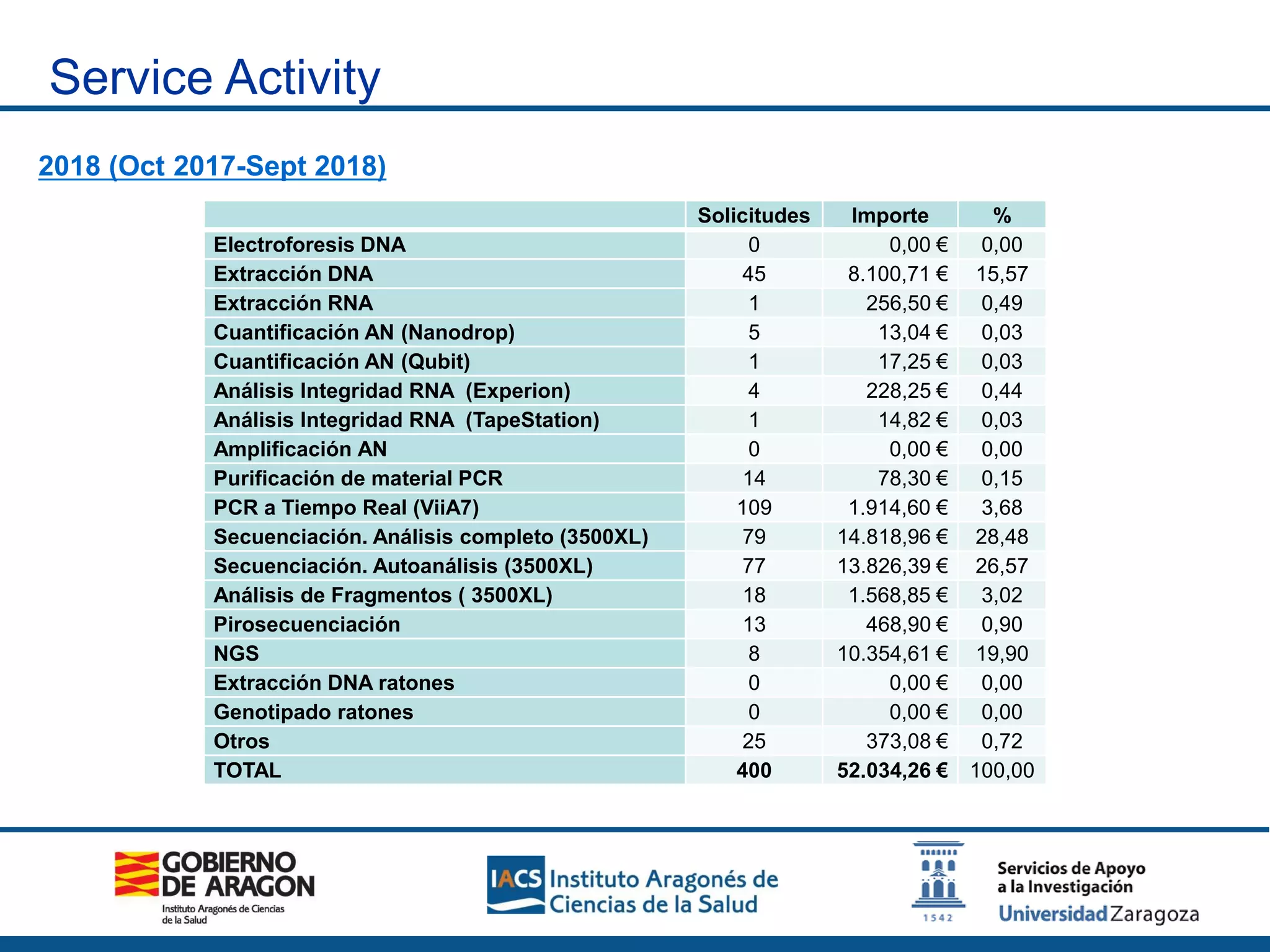
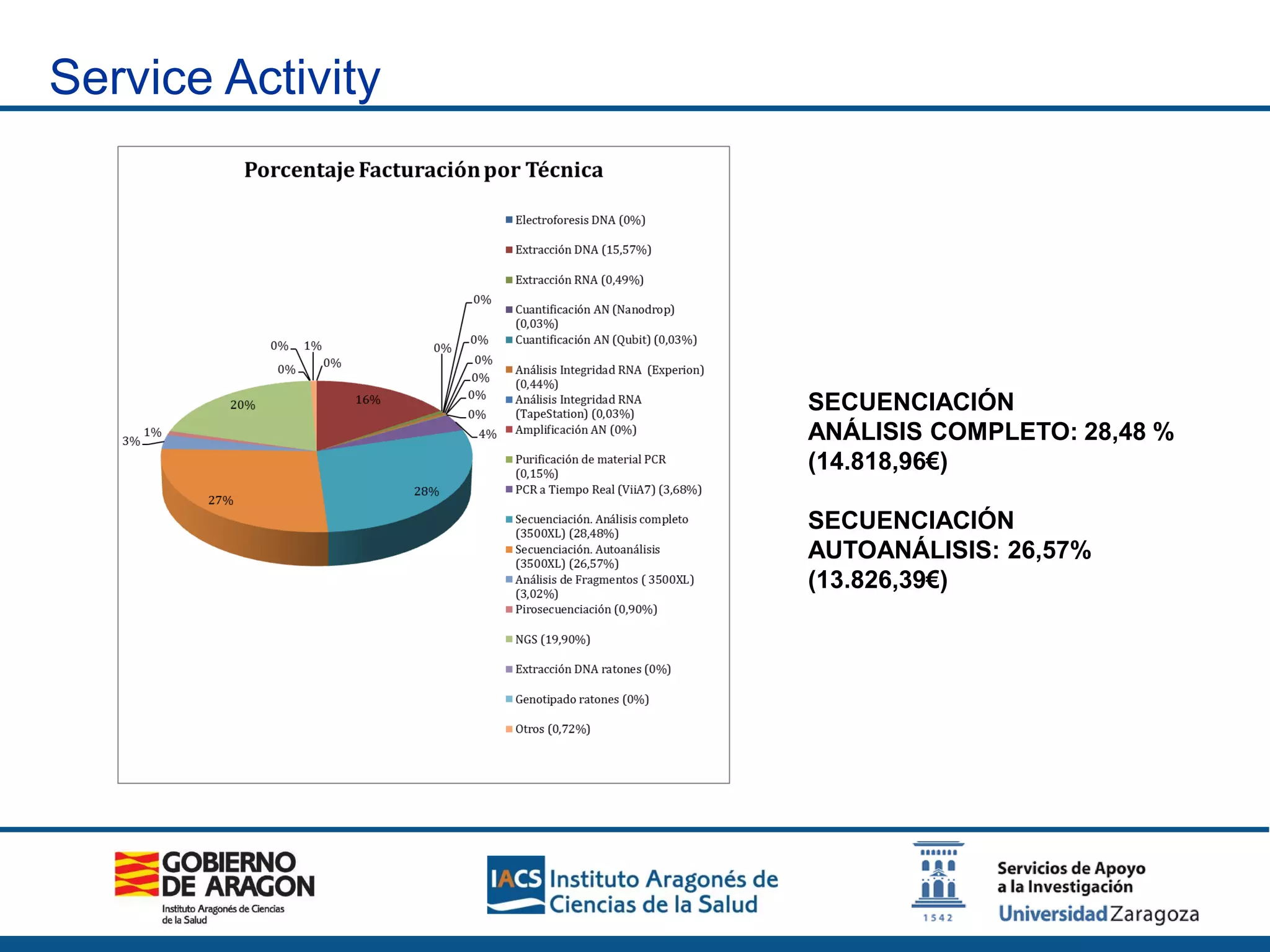
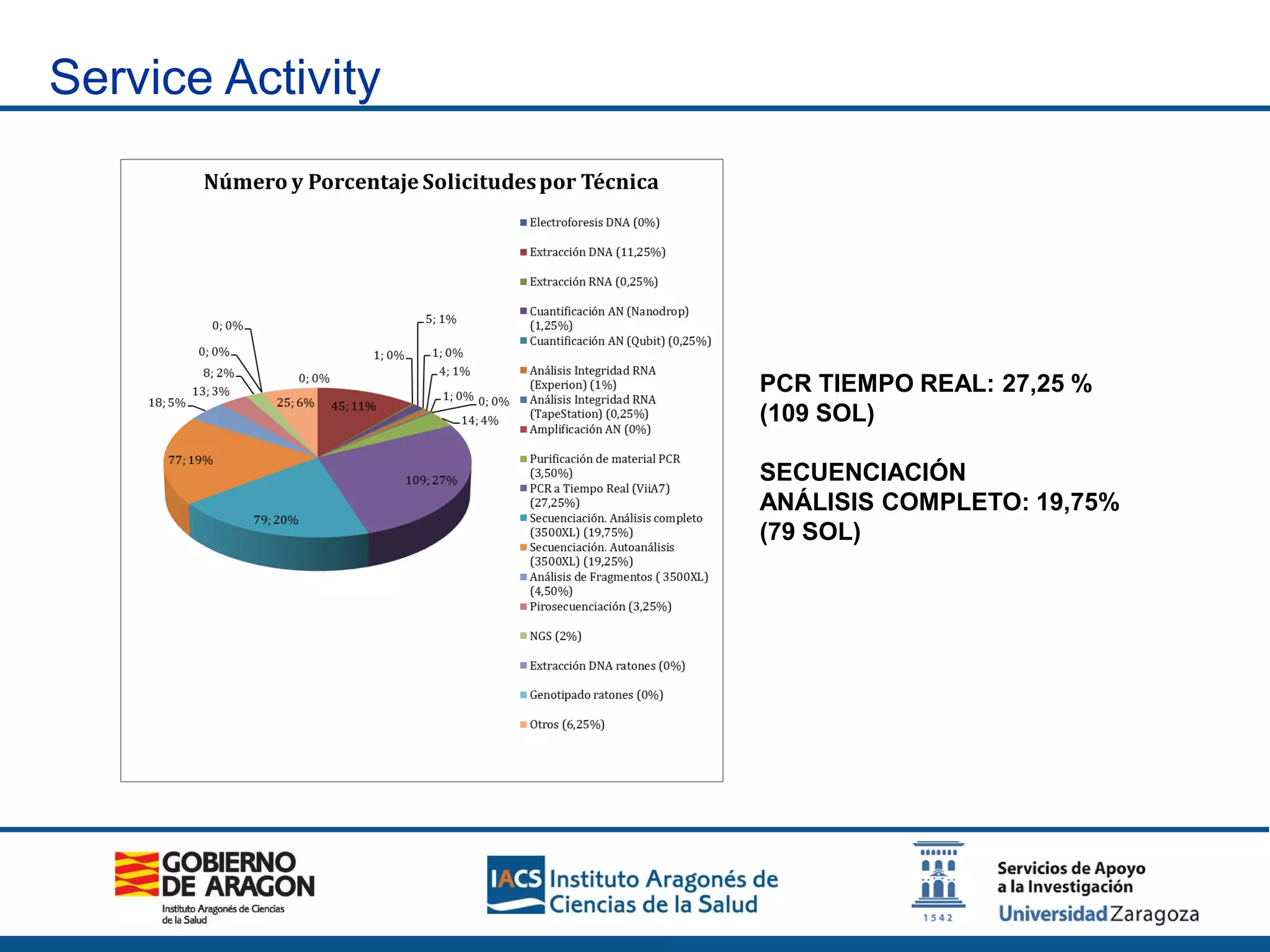
The Sequencing and Functional Genomics Unit at the Biomedical Research Center of Aragón offers a range of genomic services including real-time PCR, sequencing, and genotyping for various organisms. Key projects involve research on hereditary diseases, colorectal cancer prevention, and microbiome studies. The unit utilizes advanced equipment for DNA/RNA extraction, sequencing, and analysis to support biomedical research.































































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)











