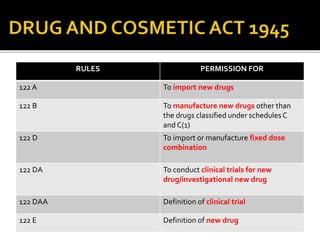
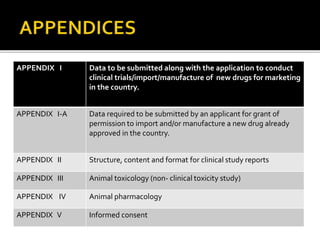
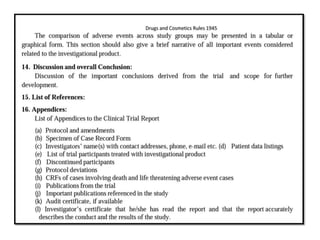
1) The document outlines guidelines for conducting clinical trials and obtaining permission to import or manufacture new drugs in India.
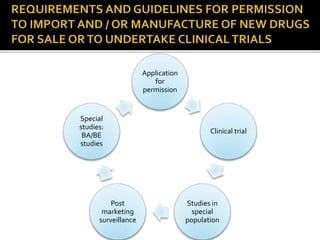
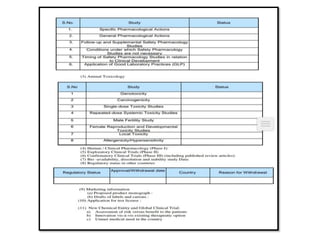
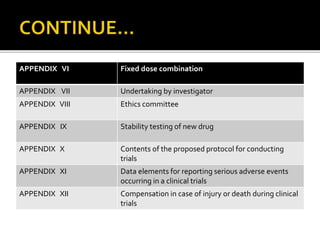
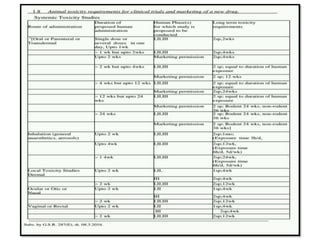
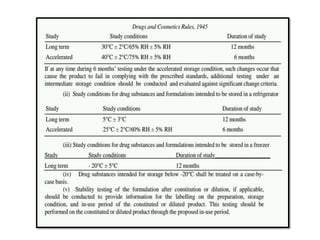
2) It discusses requirements for various types of applications and studies required including animal pharmacology/toxicology, clinical trial phases, special populations, and post-marketing surveillance.
3) The guidelines specify responsibilities of sponsors, investigators, and ethics committees in clinical trials and informed consent requirements.


































































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


