Downloaded 45 times












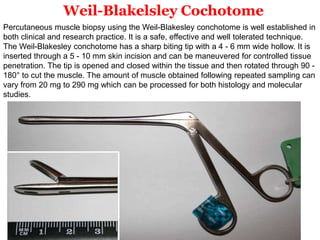




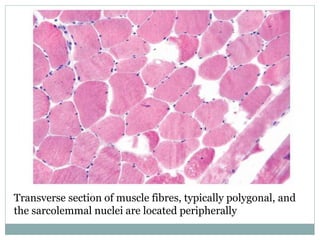








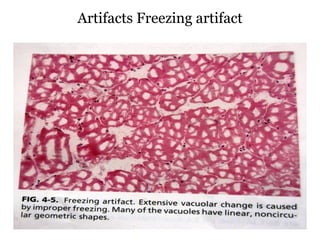

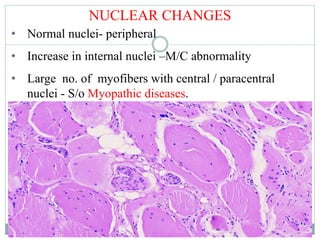

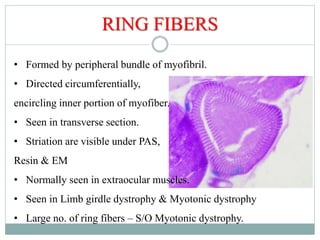











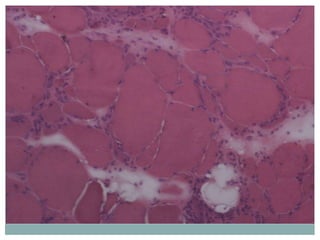















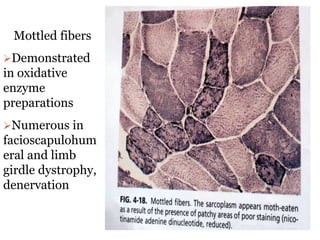






























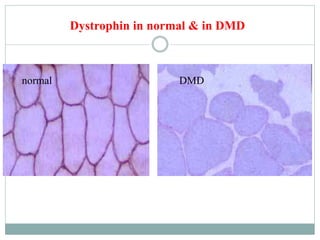




















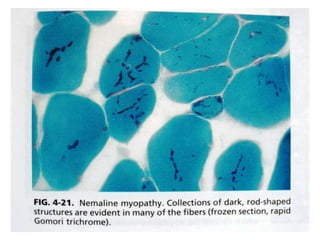










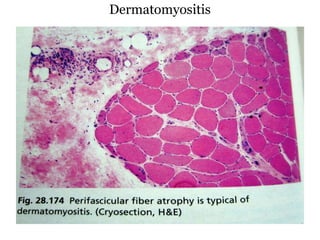



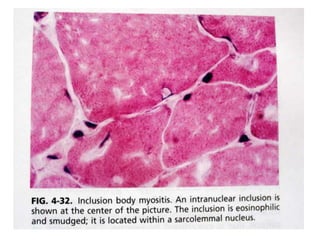
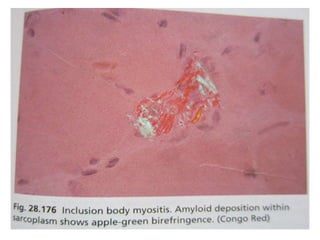









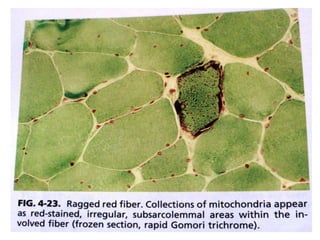


















This document provides information about muscle biopsy techniques and interpretation. It discusses how muscle biopsy can help diagnose muscle diseases based on microscopic examination. Key points include: - Muscle biopsy is useful for diagnosing muscle diseases based on histological abnormalities seen on examination of muscle tissue samples under a microscope. - The document outlines proper techniques for obtaining and preparing muscle biopsy specimens to ensure representative samples for analysis. - A variety of histological stains can be used on samples to identify abnormalities in muscle fiber size, shape, structure and histochemistry that are characteristic of different muscle diseases. - Common histopathological findings examined include changes in fiber size, fiber structure, fiber necrosis, regeneration, inflammation and histochemical profiles









































































