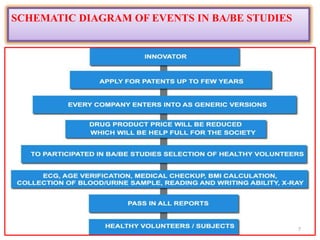
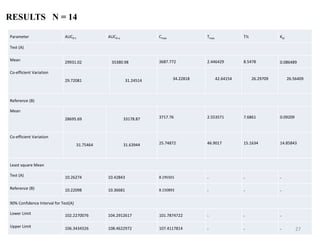
The document presents a study on the bioavailability and bioequivalence of fenofibrate SR 10 mg capsules in healthy male volunteers, comparing a generic formulation with a standard product. The results indicate that the pharmacokinetic parameters fall within acceptable limits for bioequivalence, confirming that the test drug is bioequivalent to the reference drug. This research highlights the importance of bioavailability and bioequivalence in the generic drug market to help reduce healthcare costs.






















![MEAN GRAPH OF FENOFIBRATE
28
0.000
1000.000
2000.000
3000.000
4000.000
5000.000
6000.000
7000.000
Concentration
(ng/mL)
Time in hrs
Mean Plasma Concentration Vs Time Profile
[Linear Scale, Fenofibrate 10mg, N=14]
Reference
Test](https://image.slidesharecdn.com/project-240705080812-166687d3/85/project-about-pharma-data-and-related-research-28-320.jpg)


















































