










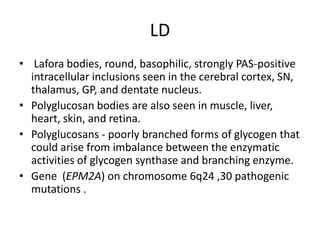

























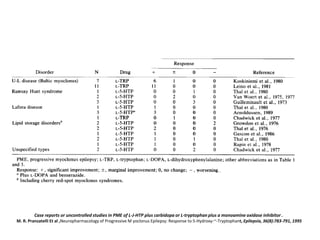




Progressive myoclonus epilepsies are characterized by myoclonic seizures, tonic-clonic seizures, and progressive neurological dysfunction including ataxia and dementia. The document discusses several specific causes of progressive myoclonus epilepsy including Unverricht-Lundborg disease, Lafora's disease, myoclonus epilepsy with ragged-red fibers, sialidoses, neuronal ceroid lipofuscinoses, and others. It provides details on clinical features, pathogenesis, diagnosis, and genetic basis for several of these conditions.























































































