Download as PDF, PPTX















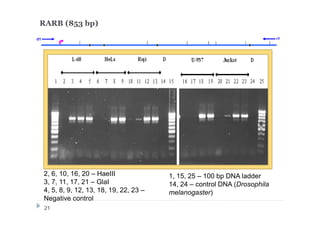
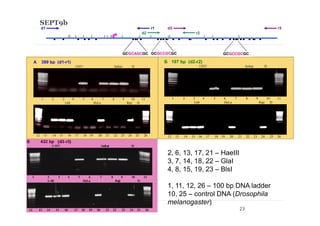
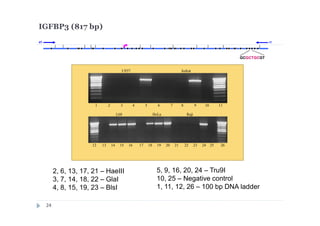
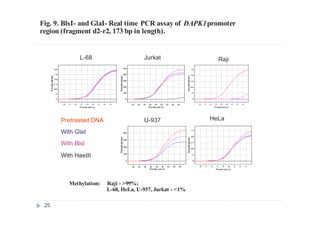
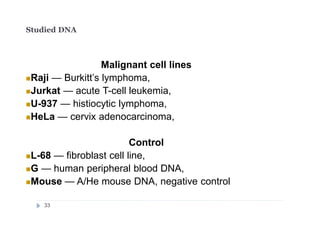
![DNA preparations from five human cell lines:
L-68 (control, lung fibroblast), HeLa (cerbix adenocarcinoma),
Raji (Burkitt’s lymphoma), U-937 (histiocystic lymphoma)
and Jurkat (acute T-cell leukemia) have been treated separately with:
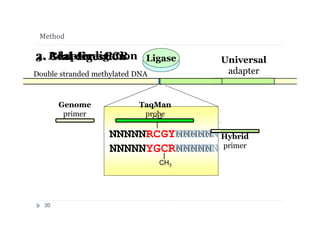
1) Restriction enzyme with recognition site in studied region
(HaeIII for CEPBD, RASSF1A and SEPT9b; FatI for RARB), positive
control;
2) GlaI (recognizes 5'-Pu(5mC)GPy-3' [2]);
3) BlsI (recognizes 5'-GCNGC-3' if at least two 5-methylcytosines (N isn't
considering) are present in both DNA strands [3]);
4) no added enzyme, negative control.
After incubation 4 reaction mixtures have been used as a DNA template for
PCR. DNA from Drosophila melanogaster at the same concentration has
been used as a negative PCR control.
Protocol of BlsI- and GlaI- PCR assay
18](https://image.slidesharecdn.com/presentation041013-140520162844-phpapp01/85/16-05-2014-18-320.jpg)









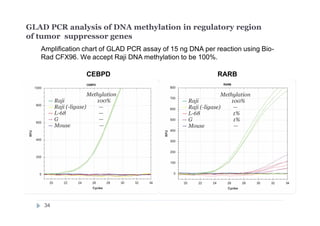
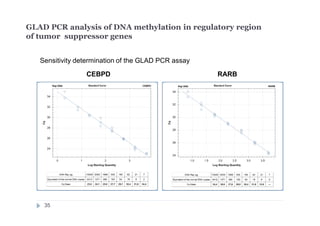
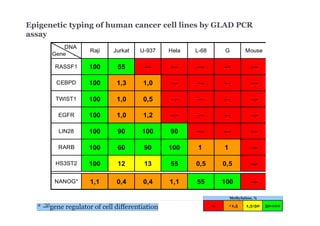


The document discusses new epigenetic tools for cancer diagnostics, particularly focusing on DNA methylation and its regulation by DNA methyltransferases like DNMT3. It compares traditional methods like bisulfite treatment with newer enzymatic approaches such as methyl-sensitive PCR assays and the recently developed glad-PCR assay for better detection of DNA methylation status in tumor suppressor genes. The findings suggest that the glad-PCR assay offers enhanced sensitivity and specificity for clinical applications in characterizing cancer-associated methylation patterns.


![Gene 151_119 (1994) [SDM of dsDNA]](https://cdn.slidesharecdn.com/ss_thumbnails/9a2273bd-32e6-4eba-b52d-2a257b547324-160607150851-thumbnail.jpg?width=640&height=640&fit=bounds)
















































































