This document discusses pharmacokinetics, specifically absorption and distribution of drugs. It covers several key topics:
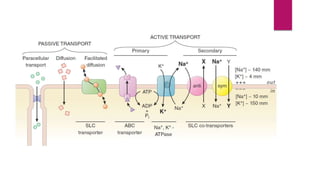
1. Modes of permeation and transport across cell membranes including passive diffusion, carrier-mediated transport, pinocytosis, and filtration.
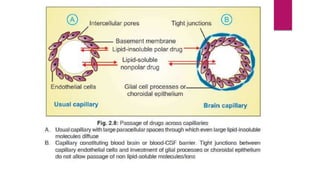
2. Factors that influence absorption including drug properties like size, ionization, and lipid solubility as well as routes of administration.
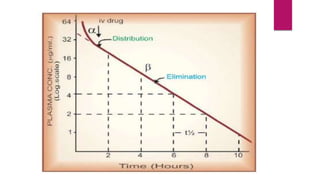
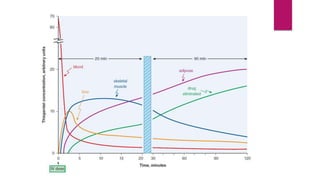
3. Distribution of drugs in the body and factors that affect it like lipid solubility, ionization, and drug-drug interactions.