Syallabus
• Pharmacokinetics
• Absorptionand bioavailability of drugs: factors affecting drug
absorption and bioavailability of drugs, difference between
bioavailability and bioequivalence.
• Distribution of drugs, apparent volume of distribution, protein binding
of drugs and their clinical significance
• Biotransformation and its types
• Excretion of drugs, plasma half- life and its significance, kinetics of
drug elimination, therapeutic drug monitoring
3.
Pharmacokinetics
• Pharmacokinetics isthe branch of pharmacology which deals with
absorption , distribution , metabolism and excretion of drugs.
• Pharmacokinetics refers to what the body does to the drugs.
Branches of Pharmacokinetics:
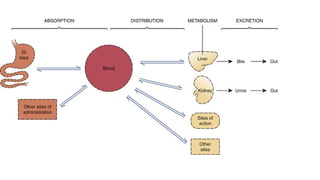
1. Absorption
2. Distribution
3. Metabolism
4. Excretion
5.
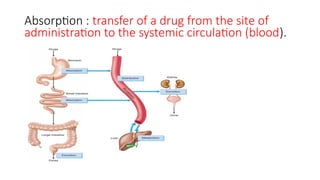
Absorption : transferof a drug from the site of
administration to the systemic circulation (blood).
6.
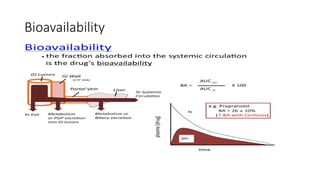
Bioavailability
( Bio: Insideliving organisms)
Availability: to make available for action
• The bioavailability of any drug is defined as the rate and extent of
drug absorbed form dosage form.
• Bioavailability refers to the extent and rate at which a drug reaches
systemic circulation or the bloodstream and becomes available at
the site of action
• it is the fraction of an administered dose of a drug that enters the
bloodstream, allowing it to exert its therapeutic effects.
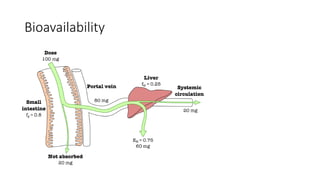
Bioavailability
Bioavailability of adrug can be defined as the
percentage amount of drug that is absorbed from a
given dosage form, reaching the systemic circulation.
• Example:
If the dose of drug administered is 500 mg, and the total amount
of that drug available in the systemic circulation is 300mg , then
what is the bioavailability?
Calculation:
Bioavailability = (total drug available in Systemic circulation) X 100 %
Dose of drug administered
= 300 x 100 %
500
= 60%
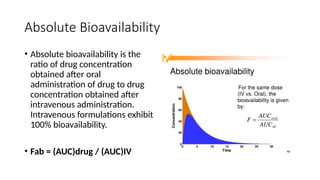
Absolute Bioavailability
• Absolutebioavailability is the
ratio of drug concentration
obtained after oral
administration of drug to drug
concentration obtained after
intravenous administration.
Intravenous formulations exhibit
100% bioavailability.
• Fab = (AUC)drug / (AUC)IV
13.
Relative Bioavailability
• Relativebioavailability is the
ratio of drug concentration
obtained after oral
administration of test drug to
that of standard of same drug
both administered orally.
• Same Drug
• F rel= (AUC) drug/(AUC)
standard
14.
• The absolutebioavailability then is the dose-corrected
area under the curve (AUC) non-intravenous divided by AUC
intravenous. The equation for calculating the absolute bioavailability
(denoted by the letter f or, if expressed in percent, by F), of a drug
administered orally (tablet) is given below:
15.
• Bioavailability ofDrug administered by Intravenous Route (IV) is
100%.
• Bioavailability of drug injected i.v. is 100%, but is frequently lower
after oral ingestion because—
(a) the drug may be incompletely absorbed.
(b) the absorbed drug may undergo first pass metabolism in the
intestinal wall/liver or be excreted in bile
Factors affecting DrugAbsorption
1. Physicochemical properties of the drug:
a. Physical state: Liquid form of the drug is better absorbed than solid formulations.
b. Lipid-soluble and unionized form of the drug is better absorbed than wate rsoluble and ionized
form
c. Particle size: Drugs with smaller particle size are absorbed better than larger ones, e.g. microfine
aspirin, digoxin and griseofulvin are well absorbed from the gut and produce better effects. Some
of the anthelmintics have larger particle size. They are poorly absorbed through gastrointestinal
(GI) tract, hence they produce better effect on gut helminths.
d. Disintegration time: It is the time taken for the formulation (tablet or capsule) to break up into
small particles and its variation may affect the bioavailability.
e. Dissolution time: It is the time taken for the particles to go into solution. Shorter the time, better
is the absorption.
f. Formulations: Pharmacologically inert substances like lactose, starch, calcium sulphate, gum, etc.
are added to formulations as binding agents. These are not totally inert and may affect the
absorption of drugs, e.g. calcium reduces the absorption of tetracyclines.
18.
Factors affecting DrugAbsorption
2. Route of drug administration: A drug administered by intravenous
route bypasses the process of absorption as it directly enters the
circulation. Some drugs are highly polar compounds, ionize in solution
and are not absorbed through GI tract, hence are given parenterally,
e.g. gentamicin. Drugs like insulin are administered parenterally
because they are degraded in the GI tract on oral administration
3. pH and ionization: Strongly acidic (heparin) and strongly basic
(aminoglycosides) drugs usually remain ionized at all pH, hence they
are poorly absorbed
19.
Factors affecting DrugAbsorption
• Food: Presence of food in the stomach can affect the absorption of some drugs. Food
decreases the absorption of rifampicin, levodopa, etc., hence they should be taken on an
empty stomach for better effect. Milk and milk products decrease the absorption of
tetracyclines. Fatty meal increases the absorption of griseofulvin.
• Presence of other drugs: Concurrent administration of two or more drugs may affect their
absorption, e.g. ascorbic acid increases the absorption of oral iron. Antacids reduce the
absorption of tetracyclines.
• Area of the absorbing surface: Normally, drugs are better absorbed in small intestine because
of a larger surface area. Resection of the gut decreases absorption of drugs due to a reduced
surface area
• Gastrointestinal and other diseases: In gastroenteritis, there is increased peristaltic movement
that decreases drug absorption. In achlorhydria, absorption of iron from the gut is reduced. In
congestive cardiac failure, there is GI mucosal oedema that reduces absorption of drugs.
20.
Factors Affecting Bioavailability
•The factors which affect drug absorption
• Physicochemical properties of the drug,
• route of drug administration,
• pH and ionization,
• food, presence of other drugs,
• area of absorbing surface,
• GI and other diseases) also affect bioavailability of a drug
• Other factors that affect the bioavailability of a drug are discussed as
follows:
21.
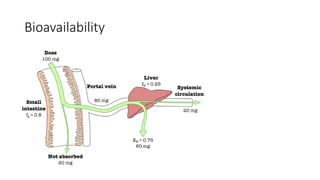
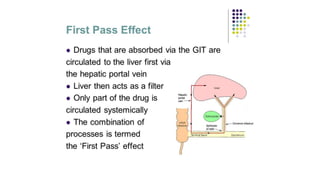
1. First-pass metabolism(First-pass effect, presystemic elimination):
When drugs are administered orally, they have to pass via gut wall n portal vein
n liver n systemic circulation . During this passage, certain drugs get
metabolized and are removed or inactivated before they reach the systemic
circulation. This process is known as first-pass metabolism. The net result is a
decreased bioavailability of the drug and diminished therapeutic response, e.g.
drugs like lignocaine (liver), isoprenaline (gut wall), etc. Consequences of high
first-pass metabolism: 1) Drugs which undergo extensive first-pass metabolism
are administered parenterally, e.g. lignocaine is administered intravenously in
ventricular arrhythmias. 2) Dose of a drug required for oral administration is
more than that given by other systemic routes, e.g. nitroglycerin
23.
2. Hepatic diseases:
Theyresult in a decrease in drug metabolism, thus increasing the
bioavailability of drugs that undergo high first-pass metabolism, e.g.
propranolol and lignocaine.
3. Enterohepatic cycling: Some drugs are excreted via bile but after
reaching the intestine they are reabsorbed --- liver—bile---intestine and
the cycle is repeated – such recycling is called enterohepatic circulation
and it increases bioavailability as well as the duration of action of the
drug, e.g. morphine and doxycycline
24.
Factors Affecting Bioavailability
First pass metabolism reduces bioavailability
Molecular weight of drug.
Drug Formulation (ease of dissolution).
(solution > suspension > capsule > tablet)
Solubility of the drug
Chemical instability in gastric pH
(Penicillin & insulin )
Factors affecting DrugAbsorption
Factors Related to Drugs:
1. Lipid water solubility
Lipid water solubility coefficient is the ratio of dissolution of drug in
lipid as compared to water. Greater the lipid water solubility
coefficient, more is the lipid solubility of the drug and greater is the
absorption. Less the coefficient, less is the lipid solubility and less is
the absorption.
28.
2. Molecular size
Smallerthe molecular size of the drug, rapid is the absorption. There
exist different processes involved in absorption for different
molecular sizes. Those with a large molecular size undergo endocytosis
or facilitated diffusion, while those with smaller molecular sizes utilize
aqueous diffusion or lipid channels.
29.
3. Particle size
Particlemay be composed either of a single molecule or more than hundred
molecules. Larger is the particle size, slower will be the diffusion and
absorption and vice versa.
4. Degree of Ionization
Different drugs are either acidic or basic and are present in ionized or
unionized form, which is given by their pKa values. In the body, the ratio of
the ionized and unionized forms depend on the pH of the medium. Acidic
drugs are unionized in the acidic medium and basic drugs are unionized in
the basic medium. Acidic drugs are better absorbed from the acidic
compartment.
30.
5. Physical Forms
•Drugs may exist as solids, liquids or gases. Gases are rapidly absorbed
than the liquids, while the liquids are rapidly absorbed than the
solids. Thus the drugs in syrup or suspension form are rapidly
absorbed than the tablets or capsules. Volatile gases used in general
anesthesia are quickly absorbed through the pulmonary route.
31.
6. Chemical Nature
•Chemical nature is responsible for the selection of the route of administration of drug
. Drugs that cannot be absorbed through the intestines are given by the
parenteral route.
• Examples include heparin which is large molecular weight, and cannot be given orally.
Simililarly, benzyl penicillin is degraded in the GIT, so is given parenterally.
• Salt forms of drugs are better absorbed than the organic compounds when given
orally. The organic compounds are given by routes other than the
oral or enteral route.
• Drugs in inorganic form are better absorbed than organic forms e.g. iron in Fe+2 is
better absorbed than Fe+3, d-tubocurarine exists in ionized form and is a quaternary
ammonium compound. Neostigmine is also a quaternary ammonium compound.
32.
7. Dosage Forms
Dosageforms affect the rate and extent of absorption. A drug can be given in the form of
tablets, capsules or transdermal packets. Injections may be aqueous or oily. This changes the rate
of absorption. Examples include nitroglycerin which when given by sublingual route, disintegrates
rapidly but stays for a shorter duration. When it is given orally, it disintegrates slowly and stays for
longer duration. When given by transdermal route, the drug can cover an even longer duration.
a. Disintegration:
• Disintegration is the breaking up of the dosage form into smaller particles. When rapid is the
disintegration, rapid will be the absorption.
b. Dissolution:
• After disintegration, the drug dissolves in the gastric juices, which is called dissolution. It is only
then that the drug can be absorbed.
• When these two processes occur rapidly, the rate of absorption increases.
33.
8. Formulation
• Whenthe drugs are formed, apart from the active form some inert
substances are included. These are the diluents, excipients and the
binders.
• Normally they are inert, but if they interact, they can change the
bioavailability. Examples include Na+ which can interact to decrease
the absorption.
• Atropine is required by some patients only in amounts of 0.2 to 0.6
mg.
34.
Factors Related toBody
1. Area of Absorptive Surface
• Area of absorptive surface affects oral as well as other routes. Most of
the drugs are given orally because of the large area of absorptive
surface, so that greater absorption occurs. Intestinal resection
decreases the surface area leading to a decreased absorption.
Similarly, when the topically acting drugs are applied on a large
surface area, they are better absorbed.
• Organophosphate compounds are highly lipid soluble and poisoning
can occur even by absorption through skin.
35.
2. Vascularity
More thevascularity, more is the rate and extent of absorption and vice versa.
In shock, blood supply to the GIT is less so the oral route of drug
administration is affected. The blood flow to the peripheries is decreased, so
absorption in those areas is diminished as well. Therefore, intravenous route
is preferred in case of shock.
Vasoconstrictors decrease the blood supply of an area, thus are useful to
restrict the local anesthesias so that they remain for a longer duration. Their
wash away as well as their toxic effects are decreased in this way.
Massage in intramuscular injections improves vascular supply to enhance
absorption.
36.
3. pH
• AcidicpH favors acidic drug absorption while basic pH is better for basic drugs.
4. Presence of other Substances
• Foods or drugs may interact with the drugs to alter their rate of absorption. Especially for the drugs given
orally, food can increase or decrease the absorption.
Antihyperlipidemic drugs like the statins are better absorbed when taken with the food.
Iron when given with milk has decreased absorption.
Vitamin C enhances the absorption of iron.
Phytates decrease iron absorption.
Milk decreases the absorption of tetracyclines.
Epinephrine when given with local anesthetics decreases their absorption.
Calcium salts when given with iron salts or tetracyclines interfere with their absorption
37.
5. GI Mobility
•GI mobility must be optimal for absorption of oral drugs. It should be
neither increased nor decreased which may affect the rate or extent
of absorption.
• Different diseases or drugs may alter the mobility. Diarrhea causes
rapid peristalsis, decreasing contact time and thus the extent of
absorption is affected more. Constipation affects disintegration and
dissolution so decreases motility.
38.
6. Gastric EmptyingTime
• Increasing the rate of gastric emptying and gastro-intestinal motility
increases the rate of absorption of a drug.
• Apart from the dissolution of drug and its permeation through the bio
membrane, the passage from stomach to small intestine, called as
gastric emptying,can also be a rate limiting step in absorption because
the major site of drug absorption is intestine.
39.
7. Diseases
a. Diarrhea
Decreasesabsorption because of decreased contact time.
b. Malabsorptive syndrome : disorder of digestive tract
Decreases absorption
c. Achlorhydria :the condition where the stomach lacks the ability to
produce hydrochloric acid (HCl),
• Acidic medium for acidic drugs is affected.
40.
What is Bioequivalence
•Bioequivalence refers to the similarity in the rate and extent to which the
active ingredient in a pharmaceutical product is absorbed and becomes
available at the site of action when compared to a reference product.
• Oral formulations of a drug from different manufacturers or different
batches from the same manufacturer may have the same amount of the
drug (chemically equivalent) but may not yield the same blood levels—
biologically inequivalent.
• Two preparations of a drug are considered bioequivalent when the rate
and extent of bioavailability of the active drug from them is not
significantly different under suitable test conditions.
44.
Difference Between Bioavailabilityand Bioequivalence
Definition
• Bioavailability refers to the rate and extent to which an active ingredient is absorbed and
becomes available at the site of action.
• On the other hand, bioequivalence compares the bioavailability of two formulations (typically
a generic and a brand-name drug) to ensure they are therapeutically equivalent.
Nature
• While bioavailability is concerned with the absorption, distribution, metabolism, and excretion
of a drug within the body, bioequivalence specifically assesses the similarity in rate and extent
of drug absorption between two formulations.
Studies
• Bioavailability studies often involve comparing different formulations or routes of
administration for the same drug. On the other hand, bioequivalence studies typically involve
comparing a generic drug to its brand-name counterpart under carefully controlled conditions.
45.
Significance Bioequivalence studies
•Bioavailability and bioequivalence studies are required to ensure
therapeutic equivalence between a pharmaceutically equivalent test
drug and a generic drug or reference drug.
46.
Mechanism of Drugabsorption
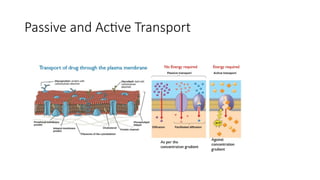
1. Passive Transport : Lipid Soluble: High conc to Low Conc
i. Passive diffusion
ii. Pore transport/ filtration
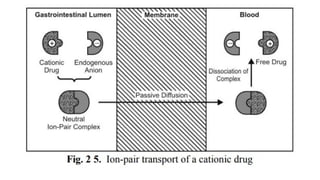
iii. Ion-pair transport
iv. Facilitated or Carrier-mediated transport
• Active Transport : Use of Energy: Low conc to high conc
• Endocytosis
Transcellular process:
A. Passivetransport process
• This type of transport process does not require energy to traverse
through the lipid bilayer.
• These are further classified as:
i. Passive diffusion
ii. Pore transport/ filtration
iii. Ion-pair transport
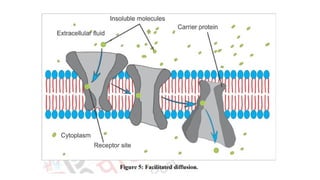
iv. Facilitated or Carrier-mediated transport
49.
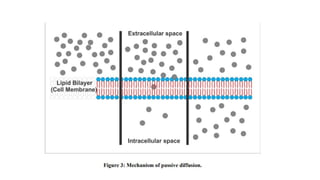
i. Passive diffusion:
•It is the movement of the drug molecule from a region of higher concentration
to a region of lower concentration.
• Concentration gradient or electrochemical gradient is considered as the driving
force for this process.
• Passive diffusion or non-ionic diffusion is considered as the major absorption
process for more than 90% of drugs.
• Movement of drugs across the membranes is a result of the kinetic energy of the
molecule.
• This process does not require any energy, so the process is known as passive
diffusion.
54.
B. Active transportprocesses
• The active transport process is a transport process in which materials
are transported against the concentration gradient i.e. from a region
of lower concentration to a higher concentration.
• It is also known as uphill transport.
• It uses energy from ATP to pull molecules from the extracellular to
the intracellular side.
• A few lipid-insoluble drugs (e.g.5-fluorouracil) are absorbed from
the GIT by this process.
55.
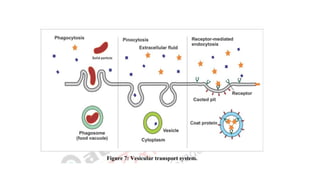
3. Vesicular orcorpuscular transport (Endocytosis)
• Like active transport this process also involves the use of energy but
differs in the case that it transports substances within the vesicles
into the cell.
• Vesicular transport can be further distinguished into two categories
such as.
a. Pinocytosis (cell drinking)
b. Phagocytosis (cell eating)
56.
a. Pinocytosis (celldrinking):
• Pinocytosis is a non-specific process whereby a substrate enters a
cell by invagination to form an intracellular vesicle.
b. Phagocytosis (cell eating):
In phagocytosis adsorptive uptake of solid substances takes place
58.
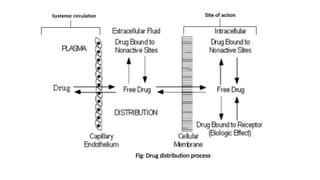
Drug Distribution
• Distributionof drugs,
• Apparent volume of distribution,
• Protein binding of drugs and their clinical significance
59.
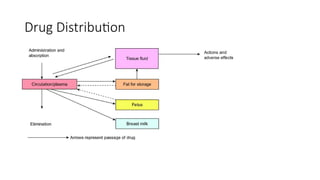
Drug Distribution
2. Drugdistribution:
• Movement of drug molecules from the systemic circulation to the site
of action/tissues.
or
• Drug Distribution is defined as the Reversible transfer of drug
between one compartment (blood) to another (extravascular tissue)
Significance of distribution:
•Distribution is the movement of drug molecules from the systemic
circulation to the site of action, the pharmacological action of a drug
depends on its concentration at the site of action, so drugs
distribution plays a significant role in:
Onset of action
Intensity of action
Duration of action
62.
Steps involved indrug distribution:
a. Permeation of Free Drug through capillary wall & Entry into the
extracellular fluid.
b. Permeation of drugs from extracellular fluid to the intracellular
fluid through the membrane of tissue cells.
64.
Drug-related factors:
1. LipidSolubility
• Lipid solubility is directly proportional to the distribution.
2. Molecular size
• Molecular size is inversely proportional to the distribution.
3. Degree of Ionization
• Only unionized drugs cross the biological membrane membrane.
65.
Factors affecting Distribution
•1. Vascularity (Blood Supply)
• Most of the blood passes through the highly perfused organs (75%)
while the remaining (25%) passes through the less perfused areas.
• Therefore, most of the drugs go first to the highly perfused areas.
They may get bound to these organs.
• Blood Flow is not equally distributed to all tissues
• High Blood Flow: Brain , Liver , Kidney : Highly Distributed.
• Low Blood Blow: Skeletal Muscle, Adipose tissue:
66.
2. Capillary Permeabilty:Small Part of Blood Vessels.
• Capillary structure
• Chemical Nature
• High Permeability : High Distribution
68.
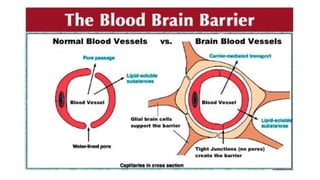
• Capillary inthe brain is highly specialized only highly lipid-soluble
drugs can cross the blood-brain barrier and distribute into neural
cells.
• All CNS-acting drugs can easily cross the blood-brain barrier.
• Following are the name of some drugs that can easily cross BBB:
• Diazepam
• Clonazepam
• Midazolam
• Morphine
69.
Body-related factors:
1. Vascularity
•Vascularity is directly proportional to drug distribution.
• They are then redistributed to the less perfused areas like the skin
and the skeletal muscles. This phenomenon is common among lipid-
soluble drugs.
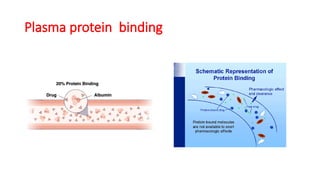
Plasma protein binding
Thephenomenon of complex formation of a drug with
protein is called protein binding of the drug.
• Acidic drugs generally binds to plasma albumin and basic
drug generally binds to α-1 acid glycoprotein.
Different types of plasma protein:
Albumin > α-1 acid glycoprotein >lipoprotein > Globulins
72.
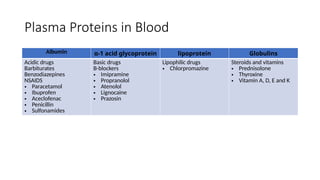
Plasma Proteins inBlood
Albumin α-1 acid glycoprotein lipoprotein Globulins
Acidic drugs
Barbiturates
Benzodiazepines
NSAIDS
• Paracetamol
• Ibuprofen
• Aceclofenac
• Penicillin
• Sulfonamides
Basic drugs
B-blockers
• Imipramine
• Propranolol
• Atenolol
• Lignocaine
• Prazosin
Lipophilic drugs
• Chlorpromazine
Steroids and vitamins
• Prednisolone
• Thyroxine
• Vitamin A, D, E and K
73.
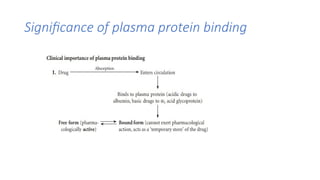
Significance of plasmaprotein binding
a. Drug absorption:
• Binding the absorbed drug to plasma protein decreases drug concentration at
desired tissue.
b. Drug distribution:
• Plasma protein binding restricts the entry of the drug to tissues.
c. Therapeutic effects:
• Only unbound or free drugs are responsible for therapeutic action.
d. Drug elimination:
• Only unbound or free drugs are capable of being eliminated.
Significance of plasmaprotein binding
2. Drugs that are highly bound to plasma proteins have a low volume of distribution.
3. Plasma protein binding delays the metabolism of drugs.
4. Bound form is not available for filtration at the glomeruli. Hence, excretion of highly plasma protein
bound drugs by filtration is delayed.
5. Highly protein bound drugs have a longer duration of action, e.g. sulphadiazine is less plasma protein
bound and has a duration of action of 6 hours, whereas sulphadoxine is highly plasma protein bound and
has a duration of action of 1 week.
6. In case of poisoning, highly plasma protein bound drugs are difficult to be removed by haemodialysis.
7. In disease states like anaemia, renal failure, chronic liver diseases, etc. plasma albumin levels are low
(hypoalbuminaemia). So, there will be a decrease in bound form and an increase in free form of the drug,
which can lead to drug toxicity.
8. Plasma protein binding can cause displacement interactions. More than one drug can bind to the same
site on plasma protein. The drug with higher affinity will displace the one having lower affinity and may
result in a sudden increase in the free concentration of the drug with lower affinity.
76.
Apparent volume ofdistribution ():
• Is a ratio of the total amount of drug in the body to the plasma
concentration of the drugs.
77.
Calculation of Apparentvolume of
Distribution
Plasma protein binding and apparent volume of distribution ():
• High plasma protein binding = small volume of distribution
• Low plasma protein binding = large volume of distribution.
78.
Significance of Volumeof Distribution
Vd is defined as the total amount of drug in the body divided by its concentration in
plasma. Thus, Vd reflects the degree to which the drug is present in extravascular tissues
rather than in the plasma.
• A drug with a high Vd tends to leave the plasma and enter other compartments in the
body, leading to low plasma concentrations. A drug with a low Vd tends to remain in
the plasma, meaning a lower dose of a drug is required to achieve a given plasma
concentration.
• Vd is dependent on both the chemical properties of a drug (e.g. highly lipid-soluble
drugs have good cell penetration, resulting in high Vd, while drugs which bind to
plasma proteins such as albumin have a reduced Vd) and patient physiology.
• Clinically, Vd is of most significance for determining an initial loading dose of an
antibiotic, assuming that successful therapy is directly linked to its plasma concentration.
79.
Significance of Volumeof Distribution
• Drugs with high molecular weight (e.g. heparin) or extensively bound to plasma protein (e.g. warfarin)
are largely restricted to the vascular compartment, hence their aVd is low.
• If aVd of a drug is about 14–16 L (0.25 mL/kg in a person weighing 70 kg), it indicates that the drug is
distributed in the ECF, e.g. gentamicin, streptomycin, etc.
• Small water-soluble molecules like ethanol are distributed in total body water – aVd is approximately 42
L. Drugs which accumulate in tissues have a volume of distribution which exceeds total body water,
■
e.g. chloroquine (13,000 L) and digoxin (500 L). Haemodialysis is not useful for removal of drugs with
large aVd in case of overdosage.
• In congestive cardiac failure, Vd of some drugs can increase due to an increase in ECF volume (e.g.
alcohol) or decrease because of reduced perfusion of tissues.
• In uraemia, the total body water can increase which increases Vd of small watersoluble drugs. Toxins
which accumulate can displace drugs from plasma protein binding sites resulting in increased
concentration of free form of drug which can leave the vascular compartment leading to an increase in
Vd.
• Fat:lean body mass ratio – highly lipid-soluble drugs get distributed to the adipose tissue. If the ratio is
high, the volume of distribution for such a drug will be higher; fat acts as a reservoir for such drugs.
80.
Biotransformation / Metabolism
•Drug metabolism is a biochemical modification of pharmaceutical
substances by living organisms usually through specialized enzymatic
activity. (Conversion of one form to another form)
Or
• It is the enzymatic conversion from one chemical form of a substance
to another.
81.
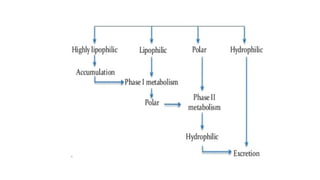
Significance of drugmetabolism:
Metabolism is an essential pharmacokinetic process, which converts
lipid-soluble and non-polar compounds to water-soluble and polar
compounds so that they are excreted by various processes.
• Normally metabolites are less toxic and easily excreted from the
body.
• Prodrugs are converted into active form and produce desired
pharmacological response after metabolism.
• Main Site : Liver
82.
Metabolim may result
1.Inactivation of Drugs: Propanolol, Lidocaine, Propanolol.
• Activation of inactive drug
2. Prodrug: (Inactive drug – Active Form)
Prodrug is an inactive substance that is converted to a drug within the body by the action of enzymes
or other chemicals.
Levodopa Metabolism Dopamine
(Prodrug) DOPA decarboxylase (Active drugs)
2. Active metabolite from Active drug :
• Allopurinol – Alloxanthine ( Active metabolite)
• Digitoxin – Digoxin
83.
Microsomal and non-microsomalenzymes:
Microsomal enzymes : Found in Liver microsomes
• Monooxygenase
• Cytochrome P-450
• Glucuronyl transferase
No microsomal enzymes: Found in Liver mitochondria
• Flavoprotein oxidase
• Amidases
• Conjugases
84.
Factors affecting drugmetabolism
Chemical factors Biological factors
a) Enzyme induction
b) Enzyme inhibition
a) Age
b) Diet
c) Gender
85.
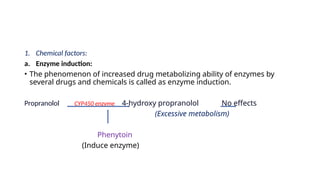
1. Chemical factors:
a.Enzyme induction:
• The phenomenon of increased drug metabolizing ability of enzymes by
several drugs and chemicals is called as enzyme induction.
Propranolol CYP450 enzyme 4-hydroxy propranolol No effects
(Excessive metabolism)
Phenytoin
(Induce enzyme)
86.
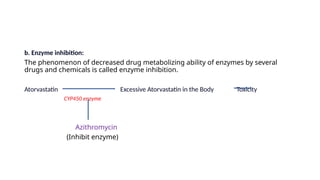
b. Enzyme inhibition:
Thephenomenon of decreased drug metabolizing ability of enzymes by several
drugs and chemicals is called enzyme inhibition.
Atorvastatin Excessive Atorvastatin in the Body Toxicity
CYP450 enzyme
Azithromycin
(Inhibit enzyme)
87.
2. Biological factors
a.Age
• The drug metabolic rate in the different age groups differs mainly due
to variations in the enzyme content, enzyme activity and
hemodynamics.
• The microsomal enzyme system is not fully developed in neonates
and infants, So, many drugs are metabolized slowly.
For e.g.: caffeine has a half-life of 4 days in neonates in comparison to 4 hr in
adults.
88.
b. Diet:
• Theenzyme content and activity is altered by a number of
dietary components.
E.g.: Grapefruit inhibits metabolism of many drugs and improve
their oral bioavailability.
c. Gender:
• Gender related differences in the rate of metabolism may be
due to sex hormones.
E.g.: In humans, women metabolize benzodiazepines slowly than
men.
89.
3. Physicochemical propertiesof the drug
• Molecular size and shape, pKa, acidity/basicity, lipophilicity
and steric and electronic characteristics of a drug influence
interaction with the active sites of enzyme and drug
metabolism process.
90.
Types of Biotransformation/Phasesof
Metabolism
Classification of drug metabolism pathways:
Phase-I or Functionalization Phase-II or Conjugation
a. Oxidation
b. Reduction
c. Hydrolysis
d. Cyclization
e. Decyclization
a. Glucuronic acid conjugation
b. Sulphate conjugation
c. Amino acid conjugation
d. Glutathione conjugation
e. Acetylation
f. Methylation
g. Alkylation
92.
Type of Biotransformation
1.Phase - I or Functionalization
Coverts the drug to a more polar (more water soluble for excretion)
• Phase-I (non-synthetic or non- conjugative phase) includes reactions
which catalyze oxidation, reduction and hydrolysis of drugs.
• In phase-I reactions, small polar functional groups like-OH, -NH2, -SH, -
COOH, etc. are either added or unmasked (if already present) on the
lipid soluble drugs so that the resulting products may undergo phase II
reactions.
• Phase-I metabolism is sometimes called a “functionalization
reaction”.
93.
• Phase-I reactionincludes:
a. Oxidation :
Addition of oxygen or –ve charged radicle to drug.
Removal of Hydrogen or +ve charged radicle to drug
Enzyme : Monooxygenase : occurs in Liver
b. Reduction : Involves the addition of hydrogen or removal of oxygen.
a. Hydrolysis : Involves the addition of water, breaking down chemical bonds.
b. Cyclization
c. Decyclization
94.
PHASE I REACTIONS
a)OXIDATION
⮦ Addition of Oxygen / negatively
charged radical or removal of Hydrogen
/ Positively charged radical
⮦ Oxidation is the main process of
metabolism
⮦ Produces unstable intermediates -
Epoxides,
Superoxides, Quinones
⮦ Oxidation – 9 types
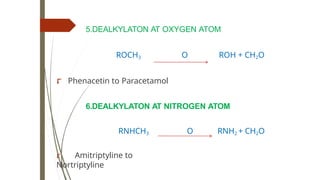
5.DEALKYLATON AT OXYGENATOM
ROCH3 O ROH + CH2O
⮦ Phenacetin to Paracetamol
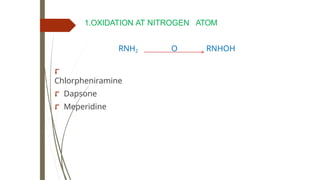
6.DEALKYLATON AT NITROGEN ATOM
RNHCH3 O RNH2 + CH2O
⮦ Amitriptyline to
Nortriptyline
100.
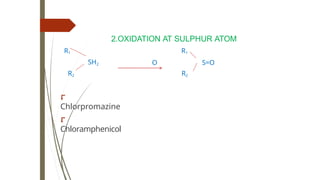
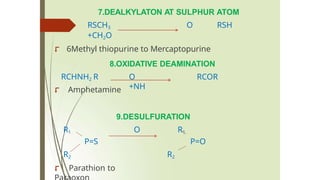
7.DEALKYLATON AT SULPHURATOM
RSCH3 O RSH
+CH2O
⮦ 6Methyl thiopurine to Mercaptopurine
RCHNH2 R
8.OXIDATIVE DEAMINATION
O RCOR
+NH
⮦ Amphetamine
9.DESULFURATION
R1 O R1
P=S P=O
R2
R2
⮦ Parathion to
101.
Main enzymes arethe Oxygenases
-
🠶MICROSOMAL MONOOXYGENASES in liver
( Cytochrome p450/CYP )- drugs
CYP( 450)s require NADPH & Oxygen
Drug Metabolizing Enzymes – 2 types
Microsomal – CYP 450, UDPGT
Non microsomal – Flavoprotein
oxidases,esterases…
b) REDUCTION
⮦ Additionof Hydrogen / positively charged radical
or
removal of Oxygen / negatively charged radical
MICROSOMAL REDUCTION by Monooxygenases
need
NADPH & cytochrome c reductase.
A.NITRO Reduction- RNo2 RNH2
⮦ Chloramphenicolto aryl amine metabolite
O
B.KETO Reduction - R-
C-R1
OH
R-CH-R1
⮦ Cortisone to
Hydrocortisone,
104.
C. AZO Reduction
⮦Prontosil to
Sulfanilamide
NON MICROSOMAL REDUCTION
⮦ Chloral hydrate to Trichloro
ethanol,
105.
c) HYDROLYSIS
•⮦ Drugis split combining with water
•⮦ Ester + water Esterases Alcohol & Acid
•⮦Microsomal hydrolysis
• Pethidine to meperidinic acid
•⮦ Non microsomal hydrolysis –
• Esterases,Amidases & Peptidases
• Atropine to Tropic acid
106.
d) CYCLIZATION
⮦ Formationof ring structure from a straight
chain compound. Eg: Proguanil
e) DE CYCLIZATION
⮦ Ring structure opened
⮦ Phenytoin, Barbiturates
107.
2. Phase –IIreaction
• Drug / phase I metabolite combines with endogenous substance
derived from carbohydrates/ proteins.
• Last step in detoxification reactions and almost always results in loss of
biological activity of a compound.
• It Involves the attachment of small polar endogenous molecules like
glucuronic acid, sulphate, methyl, amino acids, etc., to either unchanged
drugs or phase I products.
• Products called 'conjugates' are water-soluble metabolites, which are readily
excreted from the body. So Phase II metabolism is also known as conjugation
reactions.
108.
PHASE II REACTIONSCONJUGATION
/ TRANSFER
⮦ Drug / phase I metabolite combines
with endogenous substance derived
from carbohydrates/ proteins.
⮦ covalent bond formation between functional
group of drug & endogenous substrate
⮦ Endogenous-Glucuronic acid,Amino
acids, Sulfates,Acetates,Glutathione
⮦ Represent terminal inactivation – True
detoxification
reactions.
109.
• Phase-II reactionincludes:
a. Glucuronic acid conjugation
b. Sulphate conjugation
c. Amino acid conjugation
d. Glutathione conjugation
e. Acetylation
f. Methylation
g. Alkylation
1.CONJUGATION WITH GLUCURONIC
ACID
⮦UDP glucuronyl transferases
⮦ Conjugates with OH & COOH are conjugated
with glucuronic acid derived from glucose
Drug + UDPGA Microsomal
Glucuronyl
transferase
Drug glucuronide + UDP
⮦ Drugs -
Aspirin,Paracetamol,PABA,
Metronidazole,Morphine,
112.
⮦ ↑Mol.weight –favours biliary
excretion
⮦ Drug glucuronides excreted in bile are
hydrolyzed by intestinal microfloral enzymes -
parent drug released - reabsorbed into
systemic circulation-
↓excretion duration
↑ of action
- Oral contraceptives, Phenolphthalein
⮦ Endogenous substrates -
Steroid,Thyroxine,Bilirubin
113.
2. ACETYLATION
⮦ Drugswith Amino or Hydrazine
groups -
INH,PAS,Hydralazine,Sulfonamides
Procainamide,Dapsone. ( Code - SHIP)
R-NHCOCH3
⮦ R-NH2 N Acetyl
transferase
Acetyl CoA
⮦ Genetic polymorphism
⮦ Acetylation- Rapid /
Slow
4. Excretion:
• Excretionof drugs,
• Plasma half- life and its significance
• kinetics of drug elimination
• Therapeutic drug monitoring
120.
4. Excretion:
• Removalof the drug and its metabolite from the body is called
excretion.
Route of excretion:
• The main route of drugs and drug metabolites excretion is kidney/urine.
• Second most common route of drugs and drug metabolites excretion is
faeces/stool.
• Volatile and gaseous substances are eliminated via expired air.
• There are many different routes of excretion, including urine, bile,
sweat, saliva, tears, milk, and stool
121.
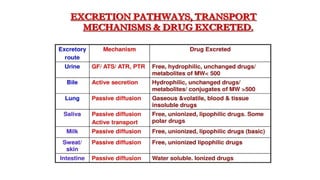
Routes of Excretion
MainRoutes of Excretion
Renal Excretion : most common, aminoglycosides
Biliary Excretion :Erythromycin, ampicillin,
rifampicin, tetracycline
Minor Routes of Excretion
Pulmonary excretion (Exhalation): Volatile (alcohol)
Salivary excretion: lithium, heavy metals
Mammary excretion via milk. : more lipid
soluble/less protrin bound
Skin / Dermal excretion via sweat.
122.
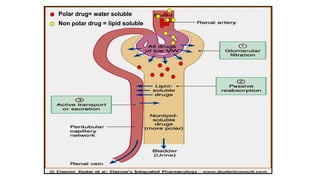
Renal Excretion
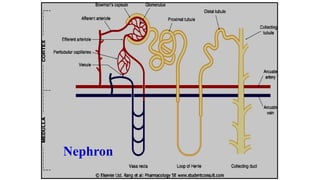
Structure ofkidney
The structure unit of kidney is nephron
That consists of :
• Glomerulus
• Proximal convoluted tubules
• Loop of Henle
• Distal convoluted tubules
• Collecting ducts
Renal Excretion includes
Theprinciple processes that determine the
urinary excretion of drugs are:
• Glomerular filtration.
• Passive tubular reabsorption.
• Active tubular secretion.
125.
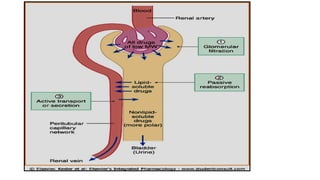
Glomerular filtration (GFR):
Depends upon renal blood flow (600 ml/min)
Glomerular filtration rate (GFR) is about 20% of
renal blood flow = 125 ml/min.
Glomerular filtration occurs to:
• Low molecular weight drugs
• Only free drugs (unbound to plasma proteins)
are filtered while bound drugs are not filtered.
126.
Active tubular secretion:
•occurs mainly in proximal tubules; increases
drug concentration in tubular lumen.
• organic anionic and cationic transporters
mediate active secretion of anionic and
cationic drugs.
• can transport drugs against conc. gradients.
Penicillin is an example of actively secreted
drug.
127.
Passive tubular re-absorption
•In distal convoluted tubules & collecting ducts.
• Passive diffusion of unionized, lipophilic drugs
• Lipophilic drugs can be reabsorbed back from
tubular lumen to blood circulation and
excretion in urine will be low.
• Ionized drugs are poorly reabsorbed & so
urinary excretion will be high.
Urinary pH trapping(Ion trapping)
• Acidification of urine using ammonium chloride
(NH4Cl) increases excretion of basic drugs as
amphetamine.
• Alkalinization of urine using sodium bicarbonate
NaHCO3 increases excretion of acidic drugs as
aspirin.
• Ion trapping is used to enhance renal clearance of
drugs during toxicity.
131.
Renal Excretion
Drugs excretedmainly by the kidney include:
• Aminoglycosides antibiotics (as gentamycin)
• B-lactam antibiotics as penicillin
• Lithium
These drugs should be prescribed carefully in
• patients with renal disease.
• Elderly people
132.
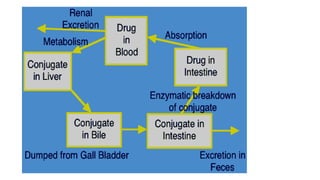
Biliary Excretion
Occurs tofew drugs that are excreted into feces.
Such drugs are secreted from the liver into bile
by active transporters, then into duodenum.
Some drugs undergo enterohepatic circulation
from intestine back into systemic blood
circulation.
133.
Enterohepatic circulation
Drugs excretedin the bile in the form of
glucouronides will be hydrolyzed in intestine
by bacterial flora liberating free drugs that can
be reabsorbed back into blood if drugs are lipid
soluble.
This prolongs the duration of action of drugs
e.g. digoxin, morphine, thyroxine.
135.
KINETICS OF ELIMINATION
Clearance(CL):
The clearance of a drug is the theoretical volume of plasma
from which the drug is completely removed in unit time.
Clearance (CL) = Rate of
eliminafion Plasma
conc. of the drug
136.
A) First-order elimination:
•The rate of elimination is proportional to the concentration (i.e., the higher
the concentration, the greater the amount of drug eliminated per unit
time).
• Here a constant fraction of drug present in the body is eliminated in unit
time
• The result is that the drug’s concentration in plasma decrease exponentially
with time.
• Drugs with first-order elimination have a characteristic half-life of elimination
that is constant regardless of the amount of drug in the body.
• The concentration of such a drug in blood will decrease by 50% for every half-
life. Most drugs in clinical use demonstrate first-order kinetics.
137.
B) Zero-order elimination:
•The term zero-order elimination implies that the rate of elimination is
constant regardless of concentration.
• A constant amount of drug is eliminated in unit time
• This occurs with drugs that saturate their elimination mechanisms at
concentrations of clinical interest.
• Such drugs do not have a constant half-life. This is typical of ethanol
and phenytoin and aspirin at high therapeutic or toxic concentration.
138.
First Order kinetics
(Linearkinetics)
Zero Order kinetics
(Non linear kinetics)
1. Constant fraction of drug is eliminated per
unit time.
2. Rate of elimination is proportional to
plasma concentration.
3. Clearance remains constant.
4. Half life remain constant.
5. Most of the drugs follow first order
kinetics.
1. Constant amount of the drug is
eliminated per unit time.
2. Rate of elimination is independant of
plasma concentration.
3. Clearance is more at low
concentrations and less at high conc.
4. Half life is less at low conc. and more
at high conc.
5. Very few drugs follow pure zero order
kinetics e.g. alcohol, warfarin,
tolbutamide
6. Any drug at high conc. (when metabolic
or elimination pathway is saturated)
May show zero order kinetics.
Plasma half-life (t½)
• Half life is the time required for the plasma concentration of a drug to fall to half
of its initial concentration.
• Is a measure of duration of action.
• Determine the dosing interval :
• It is Secondary Parameter Derived from
1. Volume of Distribution
2. Clearance
Drugs of short plasma half life
Penicillin G, tubocurarine.
Drugs of long plasma half life
Digoxin, thyroxine.
141.
Mathematically ,elimination t1/2is = ln2/k
* ln2 is the natural logarithm of 2 (or 0.693)
* k is the elimination rate constant of the drug.
i.e. the fraction of the total amount of the drug in the body which is removed
per unit time .
Eg: 2g of drug present in the body & 0.1g is eliminated every hour
then k =0.1/2=0.05 or 5% per hour .
k=CL/v, lerefore ti/2 =0.693xV/CL
142.
PRINCIPLE
The half-life ofelimination of a drug (and
its residence in the body) depends on its
clearance and its volume of distribution
t1/2 is proportional to Vd
t1/2 is inversely proportional to CL
t1/2 = 0.693 x
Vd/CL
143.
Clinical Importance ofPlasma Half-Life.
• It helps to
■ determine the duration of drug action.
■ determine the frequency of drug administration.
■ estimate the time required to reach the steady state. At steady state,
the amount of drug administered is equal to the amount of drug
eliminated in the dose interval. It takes approximately four to five half-
lives to reach the steady state during repeated administration of the
drug.
A drug is almost completely eliminated in four to five half-lives after
single administration.
144.
Significance of HalfLife
• Half Life determines Dosing Interval.
Drugs having short half life : Dosing more Frequently.
• Drugs or substances that have a shorter half-life tend to act very
quickly, but their effects wear off rapidly, meaning that they usually
need to be taken several times a day to have the same effect.
Drugs having Long Half life : Less Frequently.
• Drugs with a longer half-life may take longer to start working, but
their effects persist for longer, and they may only need to be dosed
once a day, once a week, once a month, or even less frequently.
145.
Clinical Significance:
• 1.Rate of elimination
Rate of elimination is the rate at which drug is eliminated from the
body. Certain minimum plasma levels of a drug have to be maintained
for the effect to occur.
Drugs having shorter half lives are given in frequent doses.
Drugs which are eliminated slowly, are given with less frequency. About
90-95% of the drug is eliminated after four half lives.
146.
• 2. Durationof Action
Drugs having longer half life have more duration of action and vice
versa. Ranitidine has a half life of only 2 hours, but duration of action is
about 12 hours. Although its concentration falls in the plasma but
binding to site of action is tight.
147.
• 3. Intervalbetween doses
• Drugs having short half life, the interval between the doses is kept
short and are given frequently to maintain minimum effective plasma
levels.
148.
• 4. Timefor steady state
• When the drug is given by constant intravenous infusion or given repeatedly in fixed doses at fixed
intervals, plasma concentration of drug rises gradually, and if patient is still taking the drug at fixed
intervals and doses, it reaches a peak value and then plateau is reached.
• This is because the amount of drug being administered is equal to the amount of the drug being
eliminated, which is called the steady state.
• The amount of the dug in plasma becomes constant. This can only be reached when fixed doses of drugs
are given after regular intervals. At steady state,
• elimination kinetics = assimilation kinetics.
• After about five plasma half lives the steady state is achieved. Drugs having longer half lives take longer
time to reach the steady state. Drugs having longer half lives have no immediate effect.
• For the drugs which need to be monitored, first sample is taken after the steady state has been reached.
• Lithium for bipolar disorder is an example. Plasma levels are maintained by repeated examinations
because the drug can be toxic. Its half life is about 24 hours, so the plasma levels are checked after 5 days.
149.
• 5. Timefor complete elimination
Drugs having short half lives have shorter time for complete
elimination. 90-95% of the drug is eliminated after four half lives.
150.
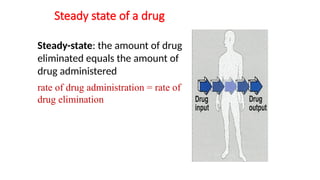
Steady state ofa drug
Steady-state: the amount of drug
eliminated equals the amount of
drug administered
rate of drug administration = rate of
drug elimination
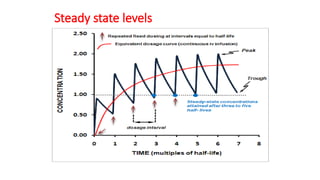
t1/2 can beused to predict how long it will take
from the start of dosing to reach steady-state
levels during multiple dosing.
No. of t1/2 Concentration achieved
(% of steady conc.)
0 100%
1 50 %
2 (50+100) 75% 3
(75+100) 87.5% 4
(87.5+100) 94%
5 (94+100) 97%
153.
How many half-liveswould be necessary to
reach steady state?
Steady state concentration is attained after 3-5
half lives.
155.
• From half-lifeestimate the duration of action for drug.
(6 x half-life) = (duration of action of the drug)
156.
Factors that mayincrease half-life (t ½ )
Decreased metabolism
• Liver disease.
• Microsomal inhibitors.
Decreased clearance
• Renal disease.
• Congestive heart failure.
High binding of drugs
• Plasma proteins.
• Tissue binding.
Enterohepatic recycling
157.
Pharmacokinetic parameters
• Volumeof distribution Vd = DOSE / C0
• Plasma clearance Cl = Kel .Vd
• Plasma half-life t1/2= 0.693 / Kel
• Bioavailability (AUC)x / (AUC)iv
Get equation of regression line; from it get Kel, C0 , and AUC
158.
Loading dose
is thelarge initial dose that is given to achieve rapid
therapeutic plasma level.
After administration of the drug, the plasma concentration
decreases due to distribution of drug to other tissues.
These doses balances the drug distribution.
This is important for drugs with long halve lives.
Loading dose =Vd x required plasma drug concentration
159.
Clinical applications ofloading dose
• A loading dose may be desirable if the time required to
attain steady state of drug is long and rapid relief is
required in the condition being treated.
• e.g. lidocaine is antiarrhythmic drug with t1/2 of
around 1-2 hours.
160.
Clinical applications ofloading dose
• Arrhythmias after myocardial infarction are life-
threatening, and one cannot wait more several hours to
achieve a therapeutic concentration.
Steady state= 3-5 X 2 hour = 6-10 hours
• Use of a loading dose of lidocaine in the coronary care
unit is standard.
161.
Maintenance doses
• arethe doses required to maintain the therapeutic level of
the drug constant or the steady state of the drug.
• These doses balance the amount of drug lost during
metabolism and clearance.
• The patient needs to take regular doses of a drug such as
amoxicillin (500 mg)/ 8 hours to maintain the therapeutic
level.
• Maintenance dose =
Clearance x required Plasma concentration
162.
CONCEPT OF THERAPEUTICDRUG MONITORING (TDM)
• TDM is based on the principle that for some drugs there is a close
relationship between the plasma level of the drug and its clinical
effect.
• The measurement of plasma level is justified only when the
information provided is of potential therapeutic benefit.
• Therapeutic drug monitoring (TDM) refers to the individualisation of
dosage by maintaining plasma or blood drug concentrations within
a target range (therapeutic range, therapeutic window).
183
163.
THERAPEUTIC DRUG MONITORING(TDM)
• Monitoring drug therapy by measuring plasma concentration of a drug is known
as therapeutic drug monitoring (TDM).
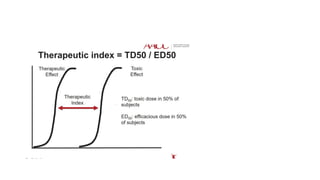
• Indications of TDM
1. Drugs with narrow therapeutic index, e.g. lithium, digoxin, phenytoin,
aminoglycosides, etc.
2. Drugs showing wide interindividual variations, e.g. tricyclic antidepressants.
3. To ascertain patient compliance.
4. For drugs whose toxicity is increased in the presence of renal failure, e.g.
aminoglycosides. 5. In patients who do not respond to therapy without any known
reason. In drug poisoning, estimation of plasma drug concentration is done.
164.
What is TDM?•
• The practice of individualized drug dosing:
• TDM is done to enhance drug efficacy and reduce the risk of toxicity
• Reserved for drugs with a well-established relationship between
blood concentration and clinical effect
• Targeted for drugs with unpredictable pharmacokinetic and
pharmacodynamics relationship with dose
• Drugs with a narrow therapeutic index
165.
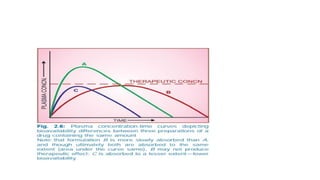
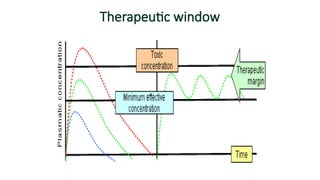
Therapeutic window:
• Isthe safe range between the
minimum therapeutic
concentration and the minimum
toxic concentration of a drug. These
data are used to determine the
acceptable range of plasma levels
when designing a dosing regimen.
e.g. therapeutic plasma conc. of
theophylline 8 mg/L and toxic effect
observed at 18 mg/L, Therapeutic
window 8-18 mg/L. Fig.1-5
Therapeutic Drug Monitoring(TDM)
• TDM is a process by which the dose of a drug is adjusted according to
its plasma concentration.
• It is done for drugs having wide variation in pharmacokinetics ,both
intra- as well as inter- individual.
• It is done for the drugs having low therapeutic index like theophylline,
lithium, antiepileptics, immuno-modulators and anti-arrhythmics etc.
• TDM is done for those whose effect cannot be easily measured (like
effect of antihypertensive drugs can be easily measured by monitoring
BP, so TDM is not used).
• TDM is not done for the drugs which are activated in the body or
produce active metabolites (Prodrugs).
169.
Indications for TDM
•Monitor patient adherence to prescribed medication
• To ensure that the patient drug concentrations are within the
therapeutic range
• Assess toxicity or adverse drug reactions
• Improve patient care through directed decontamination efforts
Factors affecting theexcretion of drugs:
1. Physiochemical properties of the drug
2.Urine pH
3. Blood flow to the kidney
4. Tissue protein binding and apparent volume of distribution
5. Biological factors
6. Disease state
173.
1. Physiochemical propertiesof the drug
• Different physiochemical properties of drugs such as molecular size,
pKa and lipid solubility affect the drug elimination process.
2. Urine pH
• Change in urine pH effect on the drug elimination process.
• Acidic drugs are eliminated more rapidly when urine pH is basic
• Basic drugs are eliminated more rapidly when urine pH is acidic
174.
3. Blood flowto the kidney
• Blood flow to the kidney is directly proportional to drug elimination.
4. Tissue protein binding and apparent volume of distribution
• It is inversely proportional to the elimination
• Drugs having high tissue protein binding and high apparent volume of
distribution eliminates slowly.
175.
5. Biological factors
•Following are the different biological factors that influence the drug
elimination process.
i. Age
ii. Gender
iii. Species
iv. Genetic makeup
176.
i. Sex –Renal excretion is 10% lower in females than in males.
ii. Age – The renal excretion in newborns is 30-40 % less in
comparison to adults.
iii. Old age – The GFR is reduced and tubular function is altered which
results in slow excretion of drugs and prolonged half-lives.
177.
6. Disease state:
•Disease state affects the drug elimination process.
• Different disease conditions such as renal impairment or hepatic
impairment compromise drug excretion.




































































































































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)







