Download to read offline



















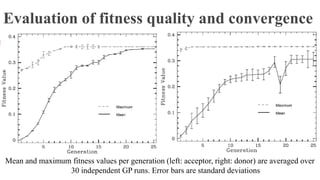











This paper proposes an evolution algorithm (FG-EA) to generate predictive features from biological sequence data for classification problems. FG-EA uses genetic programming to evolve tree-based representations of features from DNA sequences. It evaluates these features on a fitness function based on information gain before selecting high-scoring features. When applied to human and worm DNA splice site prediction, FG-EA features improved classification performance over state-of-the-art methods, demonstrating the ability of evolutionary search to discover predictive sequence features.































































