





![MICROSCOPY
• Fascicles, nodules, whorls and diffuse distribution
• Largely monomorphic spindled to epithelioid cells with few pleomorphic
cells
• Dark hyperchromatic nucleus, inconspicuous nucleoli and pale cytoplasm
• Prominent nuclear palisades, perivascular arrangement
• Mitotic figures [25/10 HPF]
• Necrosis +
• Few round cells with eccentric nucleus & eosinophilic cytoplasm](https://image.slidesharecdn.com/mpnstandmyeloidsarcoma-141102114215-conversion-gate02/85/Mpnst-and-myeloid-sarcoma-7-320.jpg)









![Final diagnosis
• MALIGNANT PERIPHERAL NERVE SHEATH TUMOR (MPNST) WITH
RHABDOMYOBLASTIC DIFFERENTIATION (MALIGNANT TRITON
TUMOR)
• [ MULTI LINEAGE DIFFERENTIATION ]
• FNCLCC GRADING : GRADE III](https://image.slidesharecdn.com/mpnstandmyeloidsarcoma-141102114215-conversion-gate02/85/Mpnst-and-myeloid-sarcoma-17-320.jpg)






![MICROSCOPIC VARIANTS
•MPNST – highly heterogenous tumor.
•MPNSTs can exhibit variable
differentiation.
[Rhabdomyoblastic, Epithelioid,
Glandular, Melanocytic, Endothelial,
Osseous, cartilaginous,
Neuro- endocrine, smooth muscle]
•EPITHELIOID MALIGNANT MPNST
•MALIGNANT TRITON TUMOR
•PERINEURIAL MPNST](https://image.slidesharecdn.com/mpnstandmyeloidsarcoma-141102114215-conversion-gate02/85/Mpnst-and-myeloid-sarcoma-24-320.jpg)




















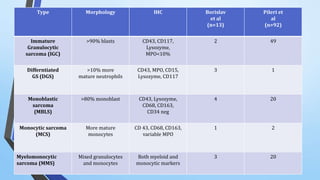








A 30-year-old female presented with a swelling in her right thigh. Biopsy and histopathological examination revealed a malignant peripheral nerve sheath tumor (MPNST) with rhabdomyoblastic differentiation. MPNSTs are rare sarcomas that arise from peripheral nerves or pre-existing neurofibromas. This case demonstrated multi-lineage differentiation, with positivity for desmin and myogenin, in addition to features of MPNST. A 40-year-old female presented with fever and weight loss. Biopsies of axillary and neck swellings revealed a granulocytic sarcoma, a type of myeloid sarcoma. Immunohistochemistry showed the
















































































