
Recommended
More Related Content
What's hot
What's hot (20)
Similar to La Fenilchetonuria
Similar to La Fenilchetonuria (20)
More from CentroMalattieRareFVG
More from CentroMalattieRareFVG (20)
La Fenilchetonuria
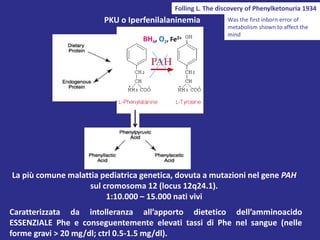
- 1. PKU o Iperfenilalaninemia La più comune malattia pediatrica genetica, dovuta a mutazioni nel gene PAH sul cromosoma 12 (locus 12q24.1). 1:10.000 – 15.000 nati vivi Caratterizzata da intolleranza all’apporto dietetico dell’amminoacido ESSENZIALE Phe e conseguentemente elevati tassi di Phe nel sangue (nelle forme gravi > 20 mg/dl; ctrl 0.5-1.5 mg/dl). BH4, O2, Fe2+ Folling L. The discovery of Phenylketonuria 1934 Was the first inborn error of metabolism shown to affect the mind
- 4. Un individuo sano ha dei valori che non superano i 2 mg/dl di Phe. Un portatore eterozigote (silente dal punto di vista clinico) ha dei valori più alti rispetto alle persone sane, perché ha maggiore difficoltà a metabolizzare la Phe (attività residua di circa il 30%), ma non supera i 2 mg/dl. Forme di Fenilchetonuria (PKU) Iperfenilalaninemia: quando i valori di Phe nel sangue, a dieta libera, sono > 2 mg/dl ma < 6 mg/dl. Anche in questi casi in Italia è previsto il trattamento (dietoterapia). Per aiutare a capire quanta Phe può tollerare un paziente nella propria dieta si distinguono 3 tipologie di PKU: PKU lieve: quando i valori di Phe nel sangue, a dieta libera, sono tra 2mg/dl e 10mg/dl e l'individuo può metabolizzare giornalmente circa 400-600 mg circa di Phe (alta tolleranza). PKU moderata: quando i valori di Phe nel sangue, a dieta libera, sono tra 10mg/dl e 20mg/dl e l'individuo può metabolizzare giornalmente 350-400 mg di Phe. PKU classica (o severa): quando i valori di Phe nel sangue, a dieta libera, sono > 20mg/dl e l'individuo metabolizza quantità di Phe inferiori a 250-350 mg.
- 5. PRINCIPALI SINTOMI: ritardo psicomotorio, disturbi nel tono muscolare, convulsioni, sonnolenza, irritabilità, anomalie del movimento, postura inusuale, ipertermia, ipersalivazione e difficoltà nella deglutizione. Si hanno complicanze più o meno severe in base alla gravità della manifestazione patologica e alla tempestività della diagnosi (NON sintomi alla nascita, Phe anomala a partire dal 4° giorno. Se non trattata è completamente IRREVERSIBILE). IMPORTANZA DELLA DIAGNOSI PRECOCE che permette l’INIZIO del trattamento prima dell’insorgenza dei sintomi. Segni in stadio avanzato: •Rush cutanei, nausea, vomito, irritabilità, eczematosi; •ridotta pigmentazione della pelle e dei capelli; •“odore di topo” di pelle, capelli e urina (fenilacetato) •Tremori, ipertono muscolare, iperreflessia tendinea
- 6. Fenilchetonuria materna o embriopatia da iperfenilalaninemia: se non trattata determina nel nascituro microcefalia, cardiopatie congenite, ritardi della crescita. Fenilchetonuria maligna (o deficit da BH4): E' una forma rara che colpisce dall'1 al 5% dei bambini fenilchetonurici. Questa forma è dovuta a un deficit del cofattore enzimatico tetraidrobiopterina (BH4). Esistono 2 tipologie di deficit da BH4: deficit della teraidropterina sintasi (PTPS) e deficit di diidropteridina reduttasi (DHPR). Fenilchetonuria benigna: nonostante si abbia la patologia non si hanno effetti (asintomatica), anche a dieta libera. Si parla poi di:
- 7. Pathways biosintetiche e rigenerative di BH4, e reazioni catalizzate da PAH: Ci sono disordini genetici associati a tutti questi steps enzimatici che causano diversi gradi di HPA e/o di squilibrata sintesi di neurotrasmettitori, causa malfunzionamento della tirosino- e della triptofano-idrossilasi, che sono idrossilasi BH4 dipendenti. (4-OH-BH4) (pterina carbinolamina deidratasi) Underhaug J, et al. Current Topics in Medicinal Chemistry (2012) 12: 2534-45.
- 8. La terapia include supplementazione di BH4, di solito accompagnata con L-DOPA e 5- idrossitriptofano. Per questa diagnosi si misurano i livelli di pteridina nelle urine e si conferma il risultato con la prova da carico orale di BH4 (20 mg/Kg), con conseguente significativa riduzione dei livelli di fenilalanina dopo 24-48 ore di trattamento. Il deficit di PTPS o di DHPR dovrebbe essere sospettato in tutti i neonati con un test di screening neonatale positivo, specialmente quando l’iperfenilalaninemia è modesta. L’HPA sensibile alla BH4 è causata da mutazioni specifiche, che codificano per proteine mutate con attività residua significativa. È caratterizzato da sintomi lievi-moderati di PKU, compresi il deficit cognitivo, le convulsioni e i disturbi comportamentali e dello sviluppo. Se non viene trattato questo deficit causa danni neurologici evidenti a 4-5 mesi di età, sebbene i segni clinici siano spesso palesi fin dalla nascita.
- 9. TRATTAMENTO Dieta con ridotto apporto di Phe, integrata con aminoacidi essenziali, vitamine e minerali al fine di riportare di Phe nella norma Somministrazione controllata di precursori (L-Dopa/carbidopa e 5-idrossitriptofano) per ripristinare la normale neurotrasmissione monoaminergica. Nelle iperfenilalaninemie lievi ha una qualche utilità il trattamento con l’analogo sintetico di BH4 (Kuvan®): i trials clinici dimostrano che circa il 40% di questi pazienti raggiungono una riduzione > del 30% dei livelli plasmatici di Phe, con conseguente aumento della tolleranza dietetica alla Phe. Si è dimostrata efficace anche in pazienti con specifiche mutazioni di PAH e con normali livelli urinari di pteridine e di attività DHPR (Database of Patients and Mutations causing BH4- responsive HPA/PKU). La BH4 è somministrata a orari precisi, in quantità relativa al peso corporeo del paziente e più volte al giorno. La terapia farmacologica, a differenza della dietoterapia, non garantisce il corretto sviluppo cerebrale.
- 10. Dieta Limitati drasticamente tutti gli alimenti ad alto contenuto proteico tra cui: aspartame (cibi “light”), alga spirulina, arachidi, frutta secca, carni altamente proteiche, caco, frumento, fagioli, ceci, fave, lenticchie, formaggi stagionati, pesce altamente proteico (tonno), frattaglie, carne di selvaggina, coniglio, legumi, farina di soia e proteine derivate dalla soia. Il fabbisogno proteico viene coperto attraverso specifici integratori proteici PRIVI di fenilalanina, che va fornito in quantità controllate in quanto amminoacido ESSENZIALE. Il GLICOMACROPEPTIDE (GMP) è una proteina derivata dal siero del formaggio, costituisce un eccellente fonte di proteine, a basso contenuto di Phe. Current guidelines: “to restore blood phenylalanine to levels as near normal as possible, as early as possible, for as long as possible, and for a lifetime in some patients”. Dieta con Phe < 500 mg/giorno (VA INIZIATA nelle prime settimane di vita) adattata a ciascun paziente per mantenere il livello plasmatico di Phe < 6 mg/dl (< 4 mg/dl nelle donne in gravidanza). Si sta valutando di abbassare questo limite. Frequente non-compliance dietetica, soprattutto negli adolescenti, deficienze nutrizionali, complicanze psicosociali, costi, imperfetto recupero neurologico dei pazienti.
- 11. PKU e gravidanza Per evitare nel feto i rischi cardiaci, di ritardo nellla crescita intrauterina, microcefalia e ritardo mentale, programmare la gravidanza e seguire una dieta controllata e rigorosa in Phe da PRIMA del concepimento (< 4 mg/dl). Questo comporta un’informazione precoce e costante per le pazienti affinchè le loro gravidanze siano programmate e seguite da un’equipe specializzata. (Iperfenilalaninemia materna o embriopatia da iperfenilalaninemia) La diagnosi di embriopatia fenilchetonurica deve anche essere considerata nel caso di bambini di donne nate prima della organizzazione dello screening di questa malattia o in un paese in cui questa indagine non esiste. Durante la gravidanza, dal 4-5° mese in poi, la tolleranza alla Phe aumenta progressivamente passando da valori, ad es di 300 mg/die a 1400 mg/die al termine della gravidanza. Questo aumento della tolleranza è dovuto all’attività epatica del feto (eterozigote) che è in grado di metabolizzare e smaltire sia la propria fenilalanina, sia quella in eccesso della mamma.
- 12. (LAT1)-transporter si lega selettivamente ai LNAAs: Val, Ile, Leu, Met, Thr, Trp, Tyr, Phe; L’aumento di Phe plasmatica nella PKU determina: 1: ridotto non-Phe LNAA uptake nel cervello causato da inibizione competitiva della Phe ; 2: aumento dell’esporto di altri LNAA dal cervello in cambio di Phe. RISULTATO: marcato aumento dell’uptake di Phe dal sangue al cervello e riduzione di quello di altri LNAA. Patogenesi della disfunzione cognitiva nella PKU
- 13. la competizione della Phe con LAT1 determina riduzioni nella sintesi cerebrale di proteine in particolare riduzione nella formazione dei poliribosomi e ridotta mielinizzazione. Patogenesi della disfunzione cognitiva nella PKU La Phe in eccesso rallenta anche lo sviluppo neuronale determinando neuroni più piccoli della media, con ridotta arborizzazione dendritica, che hanno più difficoltà a formare sinapsi con altri neuroni. la diminuzione della concentrazione cerebrale di Trp e la diminuita attività della Trp idrossilasi determina una ridotta concentrazione di serotonina cerebrale che spiega l’aumento di disturbi di ansia e depressione. RIDOTTA SINTESI di Tyr e diminuita attività della Tyr idrossilasi: minore produzione di neurotrasmettitori fondamentali nei circuiti nervosi (dopamina, adrenalina, noradrenalina) che spiega la ridotta performance neuropsicologica, tremori, atteggiamenti posturali involontari;
- 14. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. L. Adler-Abramovich, L. Vaks, Ohad Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel, E. Gazit. Nature Chemical Biology (2012) 8,701–706 . Dati recenti suggeriscono che la malattia non è solo il risultato della riduzione/assenza dell’attività enzimatica, ma anche la conseguenza della deposizione proteica: La Phe (mM) si auto-assembla in strutture nanofibrillari AMILOIDO simili Questi dati dimostrano che la PKU è strettamente relazionata alla famiglia delle malattie amiloido-dipendenti e potrebbe avere una simile eziologia.
- 15. Un tempo……Guthrie test A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants, by R. Guthrie and A. Susi, Pediatrics, 1963;32:318- 343. Diagnosi (screenings neonatali negli stati europei ed americani) Bacillus subtilis Prelievo almeno 48h dopo la nascita
- 16. Analisi mediante spettrometria di massa tandem: (utilissima perché richiede piccolissimi volumi di sangue: fino a 1.3 ml) La filosofia diagnostica è passata dal concetto “uno spot, un test, una malattia” al concetto “uno spot, un test, molte malattie”. Analisi mediante HPLC o gas cromatografia; Oggi:
- 17. quali pazienti, quale protocollo, che implicazioni a lungo termine. TERAPIE farmacologiche Trattamento con Sapropterina (analogo sintetico della 6R-BH4): Supplementazione con LNAAs (ancora controversa)
- 18. ERT per metabolizzare la Phe circolante Sono in corso studi con PEGylated PHE ammonia liasi ricombinante (rAvPAL-PEG) per i pazienti che non rispondono alla sapropterina. Il derivato con il polimero contribuisce ad eludere la rilevazione e l’eliminazione dell’enzima da parte del sistema immunitario. TERAPIE ALTERNATIVE 2) 1)
- 19. PAL: somministrazione s.c., ora si sta sviluppando una forma per os:
- 20. ONGOING: Terapia genica: studi su PAH-deficient mice hanno dimostrato che il 10% di att. enz. è sufficiente per ottenere un metabolismo normale della Phe. In un modello murino, la terapia genica diretta al fegato tramite un vettore ricombinante (rAAV: recombinant adeno-associated virus) ha permesso correzioni dell’attività enzimatica della PAH fino ad un anno, ma non permette una correzione permanente in quanto il genoma del vettore non viene integrato nel DNA degli epatociti e la rigenerazione graduale ma continua degli epatociti porta all’eliminazione del genoma del vettore. La reiniezione non è più efficace perché viene distrutto dal sistema immunitario. Il muscolo scheletrico è un organo bersaglio promettente in quanto non è soggetto a rigenerazione cellulare continua ed è più facilmente accessibile. Ma nei miociti l’espressione di PAH non porta all’idrossilazione funzionale della Phe ma vanno inseriti anche i vettori necessari anche per la produzione in loco di BH4. I risultati sono promettenti nel topo.
- 21. Phenotypic Reversion of Fair Hair upon Gene Therapy of the Phenylketonuria Mice Thöny Beat, Ding Zhaobing, Rebuffat Alexandre, and Viecelli Hiu Man. Human Gene Therapy (2014) Vol. 25, No. 7: 573-574.
- 22. Le mutazioni maggiormente patogeniche nella PKU sono associate all’aumentata tendenza della proteina al misfolding e alla degradazione ubiquitina-dipendente Questo ha stimolato lo studio di chaperones come potenziale terapeutico. Trattamento con chaperones farmacologici Nel caso di PKU, il cofattore fisiologico 6R-BH4, oltre che come co-substrato agisce come uno chaperone farmacologico di PAH, formando il complesso stabile PAH-BH4 che blocca e stabilizza PAH attraverso un cambiamento conformazionale, proteggendola dalla degradazione da parte del sistema di “controllo qualità” della cellula. Questo spiegherebbe l’efficacia del trattamento con BH4 in alcuni pazienti con specifiche mutazioni che generano un misfolding della proteina PAH (che sono quelle prevalenti) e inducono una PKU moderata. Il fenotipo responsivo però non può essere predetto dal solo genotipo (l’HPA è una condizione multifattoriale in cui fattori genetici e non- contribuiscono a inconsistenze nelle relazioni genotipo-fenotipo). Underhaug J, et al. Current Topics in Medicinal Chemistry (2012) 12: 2534-45. Circa il 40% dei pazienti con PKU lieve raggiunge una riduzione stabile >30% dei livelli plasmatici di Phe.
- 23. Riassunto dei potenziali approcci terapeutici
- 24. Futuro: Terapia personalizzata (misfolding protein) Underhaug J, et al. Current Topics in Medicinal Chemistry (2012) 12: 2534-45.
- 25. Ad oggi, il trattamento maggiormente impiegato è la dieta a basso contenuto di Phe. Il vantaggio è che può essere utilizzata per tutte le mutazioni, con risultati accettabili, poiché viene adattata a ciascun paziente monitorando i livelli di Phe. Importanza di trovare alternative terapeutiche, vista anche la possibilità delle conseguenze secondarie alla deposizione proteica Nonostante i recenti miglioramenti nella dieta (maggiore scelta di prodotti con migliori caratteristiche organolettiche), presenta ancora gli inconvenienti di una dieta che deve durare per tutta la vita (conseguenze psico-sociali) e l’esigenza di controlli periodici. Al presente, la supplementazione di BH4 rappresenta una reale opzione terapeutica per i pazienti responsivi.