



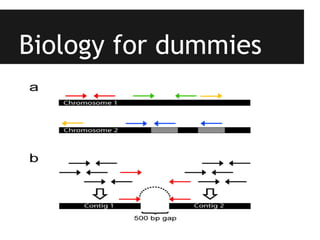


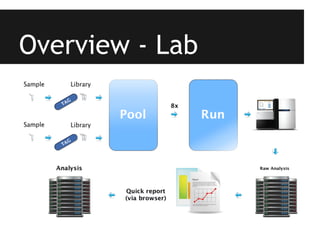




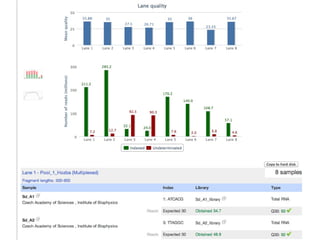












This document provides an overview of IGA Workflow, a web-based tool for managing samples in a wet lab from receipt to sequencing results. Key features include tracking samples, libraries, and pools; configuring sequencing runs; integrating analysis pipelines; and developing a genome browser to view results. The tool was created using Django and integrates technologies like Celery, Redis, PostgreSQL, and nginx to provide scalable pipeline management and optimize computational resources for analysis of hundreds of samples. Future plans include direct sample input from customers and integration of barcodes.







































































