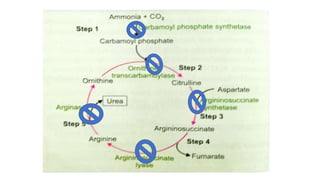
Hyperammonemia is a medical condition characterized by an abnormally elevated level of ammonia in the bloodstream. It is caused by defects in the urea cycle which is responsible for detoxifying ammonia produced from protein catabolism. Symptoms range from lethargy and vomiting to seizures and coma. Diagnosis involves tests of blood and urine amino acid and organic acid levels as well as genetic testing. Treatment focuses on restricting protein intake, supplementing with alpha-ketoacid derivatives, and administering drugs to conjugate ammonia into excretable compounds to lower blood ammonia levels.