Downloaded 220 times


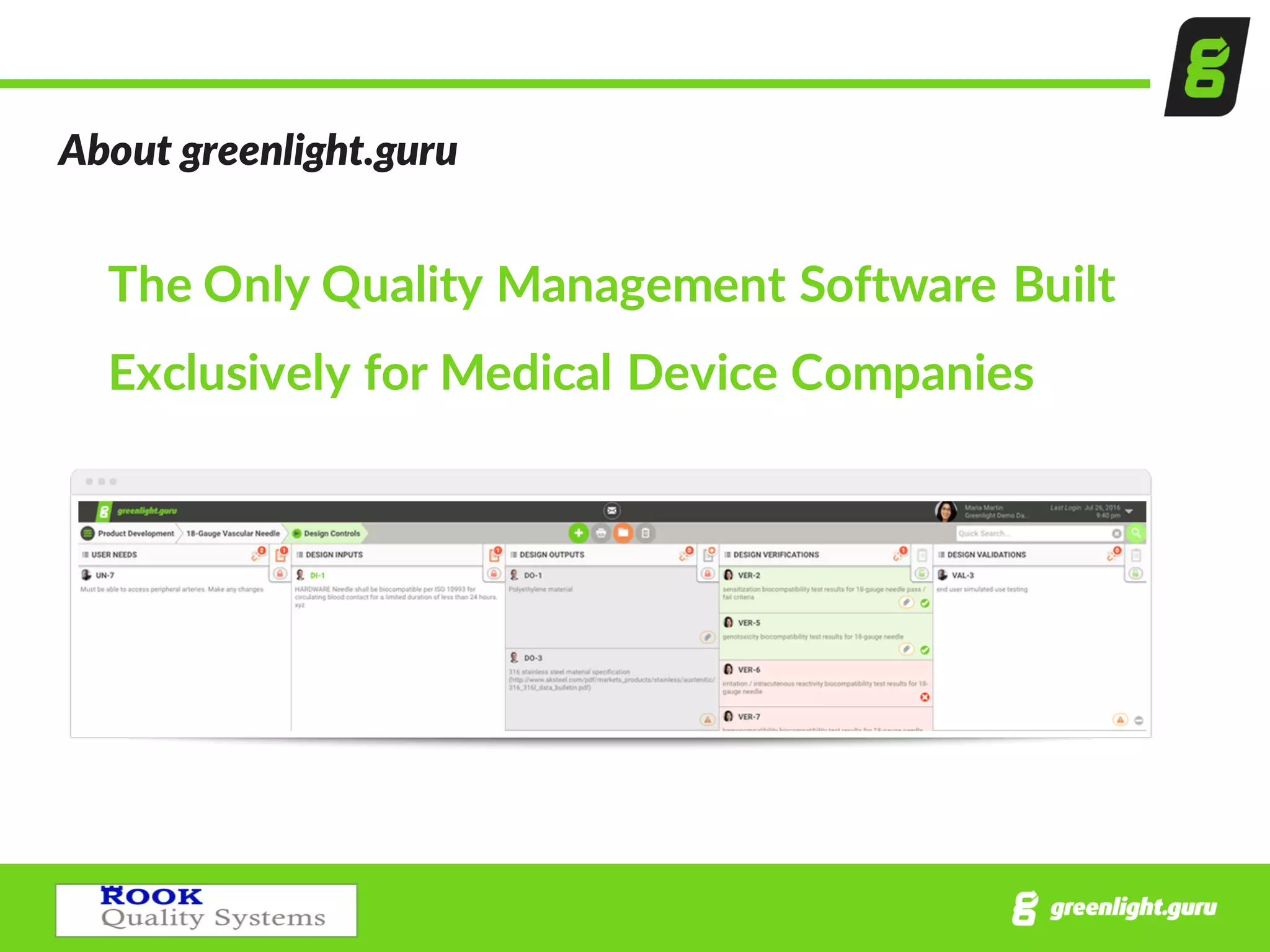






























The document outlines a comprehensive guide on conducting successful quality audits, focusing on internal audits for medical device companies. It defines the process, preparation, and best practices while emphasizing the importance of compliance with regulatory standards. Additionally, it includes details on handling audit findings, reporting, and common mistakes to avoid in the auditing process.
Overview of the presentation on performing a successful quality audit and introduction of presenter Kyle Rose, who leads a firm specialized in medical device quality systems.
Explanation of internal audits with references from FDA and ISO standards, outlining compliance and requirements for quality audits.
Internal audits are essential for monitoring procedures, ensuring product safety, effectiveness, and compliance with regulations.
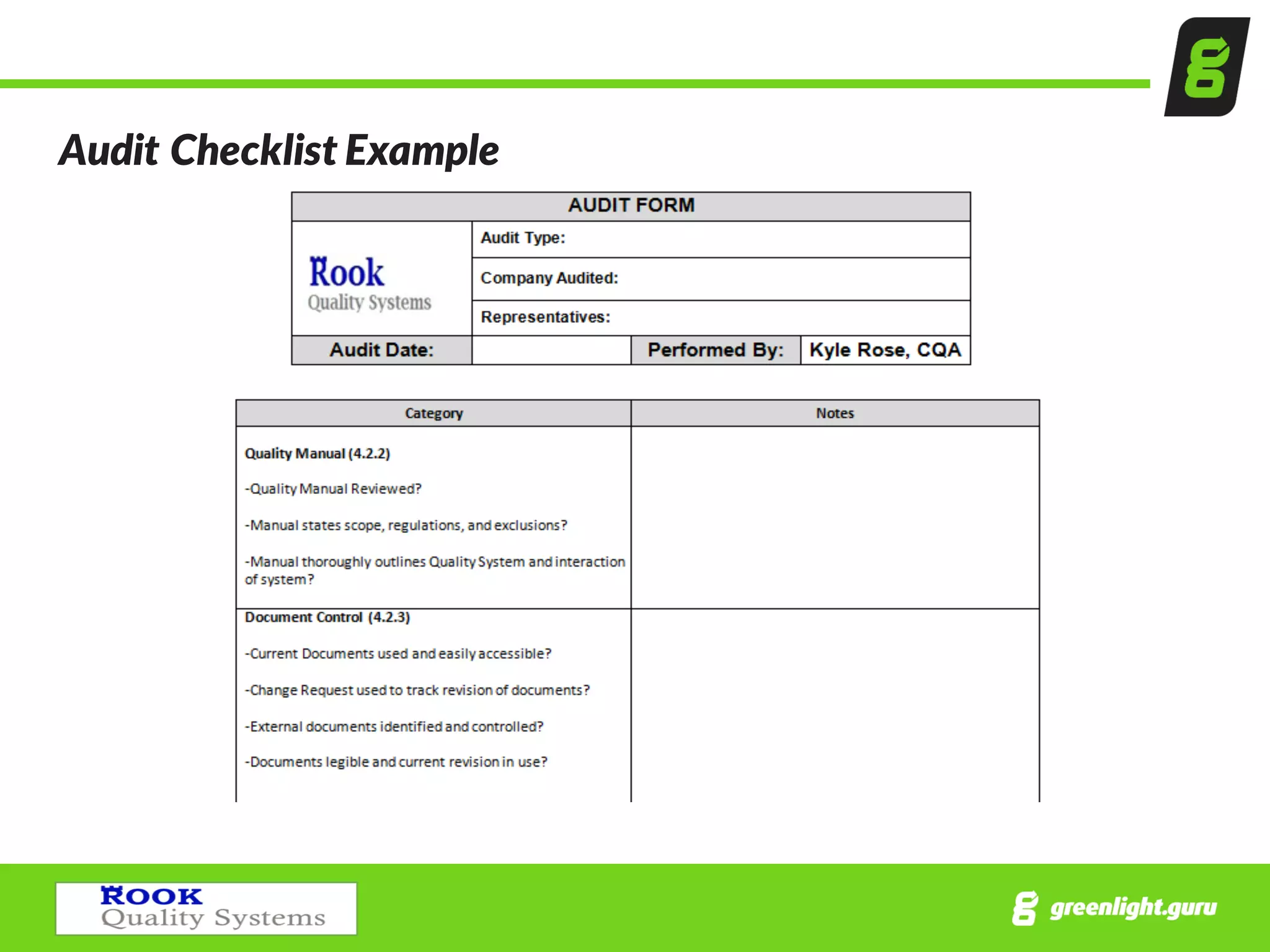
Preparation steps for internal audits including establishing procedures, creating checklists, and necessary forms.
Steps for conducting audits, including logistics, initial document reviews, and validation of design control and risk management.
Procedures for managing audit findings, including definitions, notifications, and the importance of risk-based actions.
Components of the audit report, including format, summaries, findings, CAPA assignments, and gap analysis requirements.
Highlights frequent pitfalls to avoid during internal audits, ensuring improved compliance and audit effectiveness.
Provides contact details for Rook Quality Systems for further audit-related inquiries and assistance.
Opportunity for questions and prompts to access a free QMS audit checklist to assist organizations.






















































![Iqa training -manufacturing[1]](https://cdn.slidesharecdn.com/ss_thumbnails/iqatraining-manufacturing1-191018130645-thumbnail.jpg?width=640&height=640&fit=bounds)




















































