Download to read offline

![View Article Online
Published on 05 December 2013. Downloaded by University of Texas Libraries on 18/12/2013 16:49:31.
Paper
RSC Advances
with the ability to control, stabilize, and disperse the metal
nanoparticles can greatly enhance the performance. In particular, there has been increased interest in graphene nanoplatelets (GNPs). They have been explored as durable catalyst
supports for fuel cells due to the following distinct characteristics: (1) superior conductivity and (2) strong corrosion resistance.19–22 We demonstrate here, for the rst time, that Pd–Au
nanoparticles supported on graphene exhibit enhanced electrocatalytic activity and stability for formic acid oxidation
compared to Pd supported on carbon (Pd/C).
2.
Experimental
2.1. Functionalization of graphene nanoplatelets by PDDA
(PDDA–xGNP)
All the chemicals are of analytical grade and were used as
received. The graphene nanoplatelets (xGNP) (purity $ 99.5%)
were obtained commercially from XG Sciences (USA).23,24 These
nanoplatelets were small stacks of graphene sheets, about 5–10
nm in thickness with a specic surface area of 112 m2 gÀ1.25
xGNP was functionalized with a long-chain positively charged
polyelectrolyte, poly (diallyldimethylammonium chloride)
(PDDA) (MW ¼ 200k to 350k, Sigma-Aldrich). PDDA can be
easily adsorbed onto the hydrophobic surface of xGNP via the
p–p interaction between the unsaturated C]C contaminant in
the PDDA chains22,25 and graphene planes of xGNP. Typically,
300 mg of xGNPs was dispersed in 500 mL of 0.5 wt% PDDA
aqueous solution and ultrasonicated for 3 h, which yielded a
stable dispersion of xGNP. Then, the dispersed solution of
xGNP was stirred for 24 h. Aer that, 2.5 g of KNO3 was added to
increase the binding between PDDA and xGNP surface, resulting in a highly functionalized xGNP with PDDA.21 The solution
was stirred for another 24 h, ltered, and washed with ultrapure
deionized water (18.2 MU cm, Mill-Q Corp.) to remove the free
polyelectrolyte and then dried for 3 h at 90 C in vacuum, which
is hereaer denoted as PDDA–xGNP.
2.2. Synthesis of Pd–Au/PDDA–xGNP catalysts
First, 0.9433 g of sodium citrate was dissolved in 165 mL of
water–ethylene glycol mixture solution (volume/volume ¼ 1 : 1),
and then 160 mg of functionalized PDDA–xGNP was transferred
into the above solution to obtain the sodium citrate suspension,
which was stirred and ultrasonically mixed for 2 h. 44.5 mg of
K2PdCl4 and 44.5 mg of HAuCl4$3H2O (aqueous solution containing 1 g per 100 mL) were dissolved in another 40 mL of
water–ethylene glycol solution. The sodium citrate suspension
was reuxed at 170 C in argon atmosphere for 5 min. The
catalyst precursor solution was then added drop-wise into the
heated sodium citrate suspension and the solution was diluted
by adding 40 mL of water–ethylene glycol solution (volume/
volume ¼ 1 : 1) and continued to be heated for another 2 h. The
catalyst thus obtained with 20 wt% metal loading was then
ltered, washed with water and ethanol, dried at 60 C for 12 h,
and then reduced in hydrogen at 150 C for 2 h. The as
synthesized catalyst is hereaer denoted as Pd–Au/PDDA–
xGNP. The Pd supported on PDDA–xGNP sample with 20 wt%
This journal is © The Royal Society of Chemistry 2014
Pd was prepared by the same process, but without including the
Au precursor in the synthesis, and this sample is hereaer
denoted as Pd/PDDA–xGNP. Commercial Pd/C catalyst was
obtained from Johnson Matthey.
2.3. Preparation of working electrode
The Glassy Carbon (GC) electrode was polished before each
experiment to a mirror nish with 0.05 mm alumina suspensions and rinsed thoroughly with double distilled water. The
electrode was dried in a high purity nitrogen stream. The
catalyst ink suspensions were prepared by mixing the required
amount of catalyst in 0.5% Naon solution. The mixture was
sonicated for 30 min in an ultrasonication bath and 7 mL of the
resulting catalyst ink was cast onto the surface of the GC electrode (5 mm diameter, 0.196 cm2). The modied electrode was
allowed to dry at 80 C for 5 min to obtain a uniform catalyst
lm. All electrochemical experiments were carried out at room
temperature and ambient pressure, employing 0.5 M sulphuric
acid as the electrolyte solution.
2.4. Characterization methods
X-ray diffraction (XRD) patterns were recorded with a Philips
Xpert X-ray diffractometer using Cu Ka radiation. For transmission electron microscopy (TEM) studies, the catalysts
dispersed in ethanol were placed on a copper grid and the TEM
images were collected with a JEOL 2010 TEM equipped with an
Oxford ISIS system. The operating voltage on the microscope
was 200 keV. All images were digitally recorded with a slow-scan
charge-coupled device (CCD) camera.
2.5. Electrochemical measurements
All electrochemical studies were carried out with an Autolab
PGSTAT 30 (Eco Chemie) potentiostat/galvanostat. A classical
three-electrode cell consisting of Ag/AgCl (3 M KCl) reference
electrode, a platinum plate (5 cm2) counter electrode, and a
glassy carbon working electrode (the diameter of working
electrode is 5 mm, 0.196 cm2) was used for the cyclic voltammetry (CV) studies. The CV experiments were performed with
0.5 M H2SO4 solution in the absence and presence of 0.5 M
HCOOH at a scan rate of 50 mV sÀ1. All the solutions were
prepared with ultra-pure water (Millipore, 18 MU). The electrolytes were degassed with nitrogen gas before the electrochemical measurements. All the electrochemical data were
collected at room temperature.
3.
Results and discussion
3.1. Physiochemical characterization of the catalysts
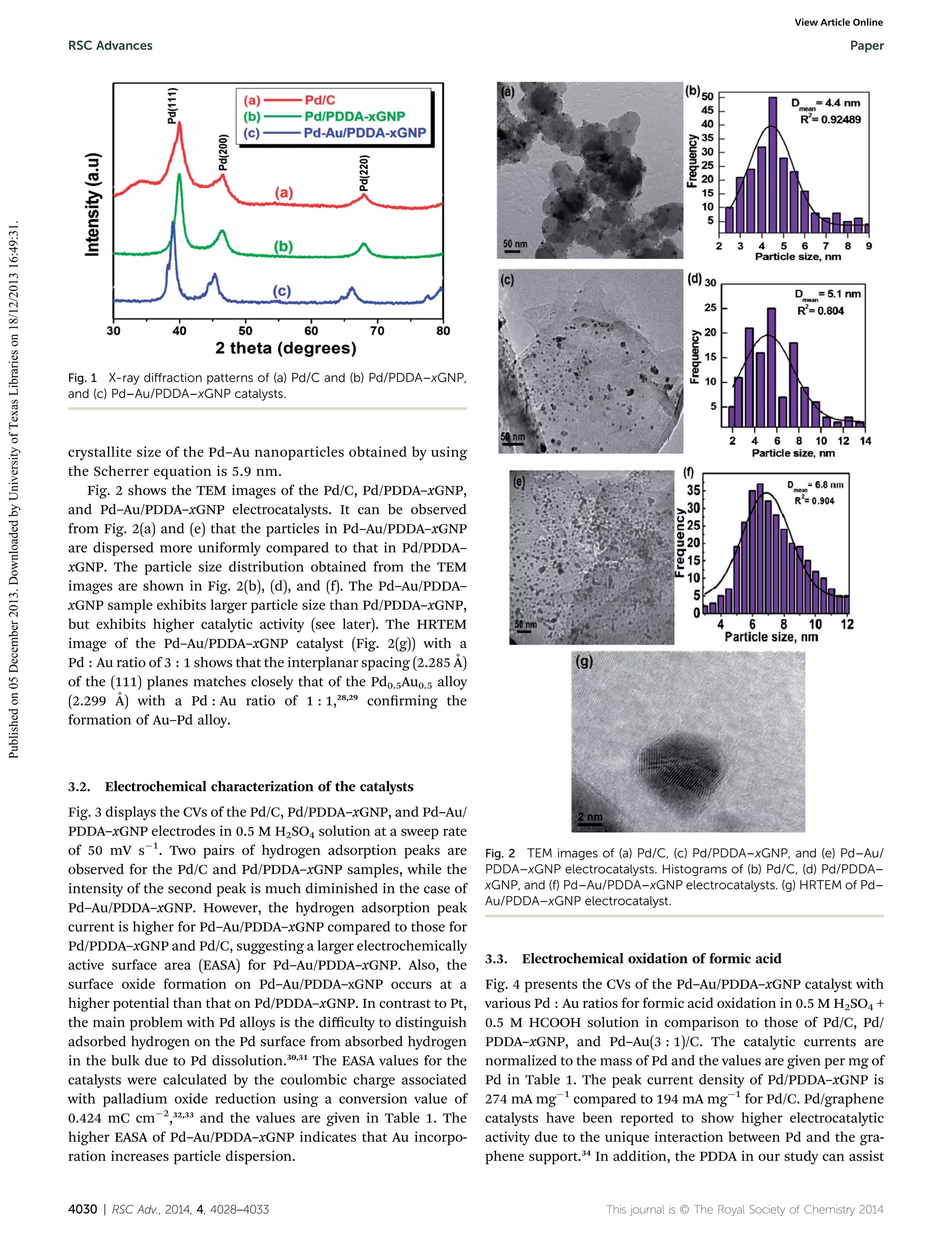
The XRD patterns of the Pd/C, Pd/PDDA–xGNP, and Pd–Au/
PDDA–xGNP samples are shown in Fig. 1. All the three
samples show reections characteristic of a face-centered
cubic (fcc) lattice, corresponding to the structures of Pd or
Pd–Au.26,27 The reections of Pd–Au (3 : 1 atomic ratio) are
shied to lower angles compared to those of Pd due to the
larger size of Au, conrming the alloying of Au with Pd. The
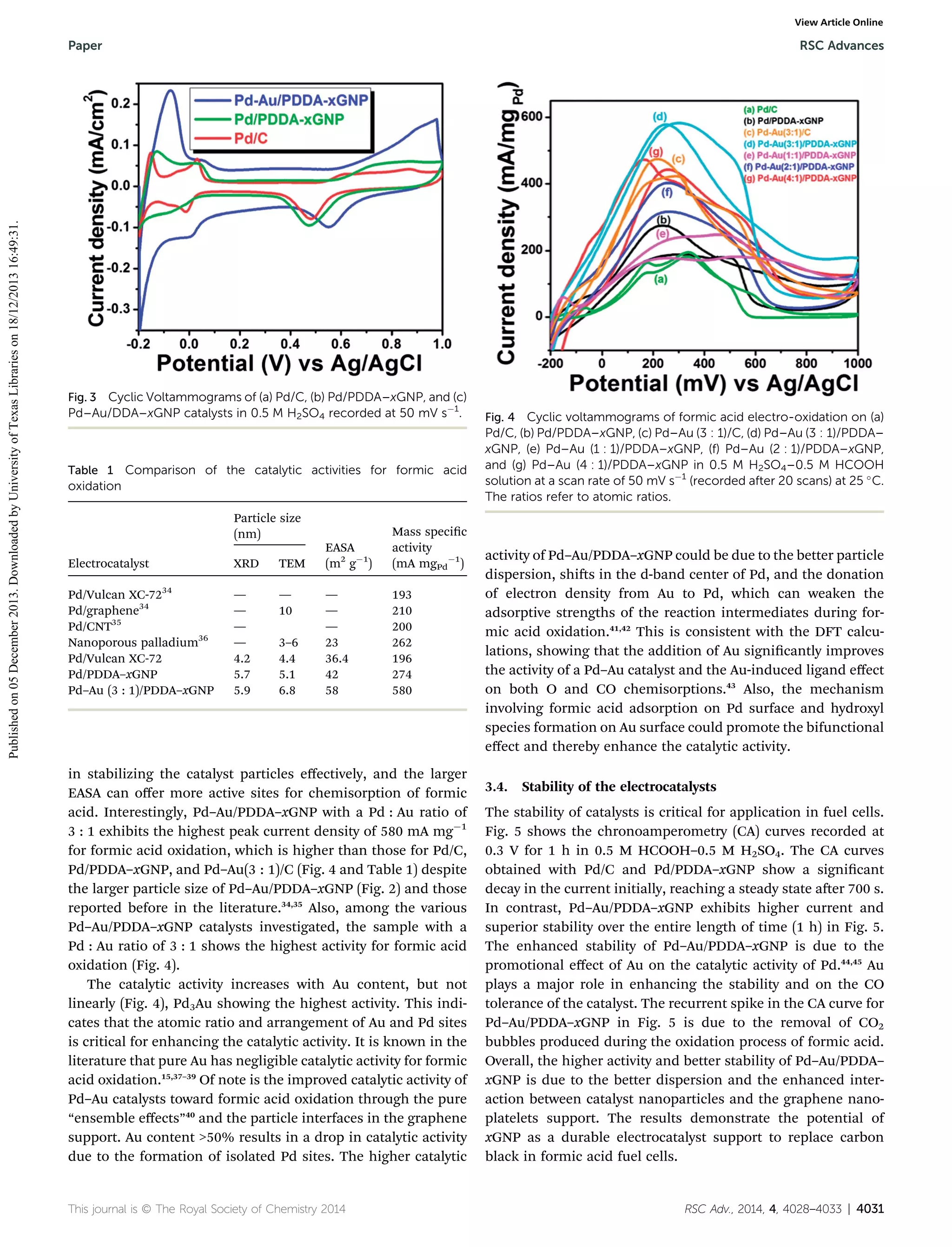
RSC Adv., 2014, 4, 4028–4033 | 4029](https://image.slidesharecdn.com/highlyactivepdandpdaunanoparticlessupportedonfunctionalizedgraphenenanoplateletsforenhancedformicaci-131218105424-phpapp01/75/Highly-active-pd-and-pd-au-nanoparticles-supported-on-functionalized-graphene-nanoplatelets-for-enhanced-formic-acid-oxidation-2-2048.jpg)
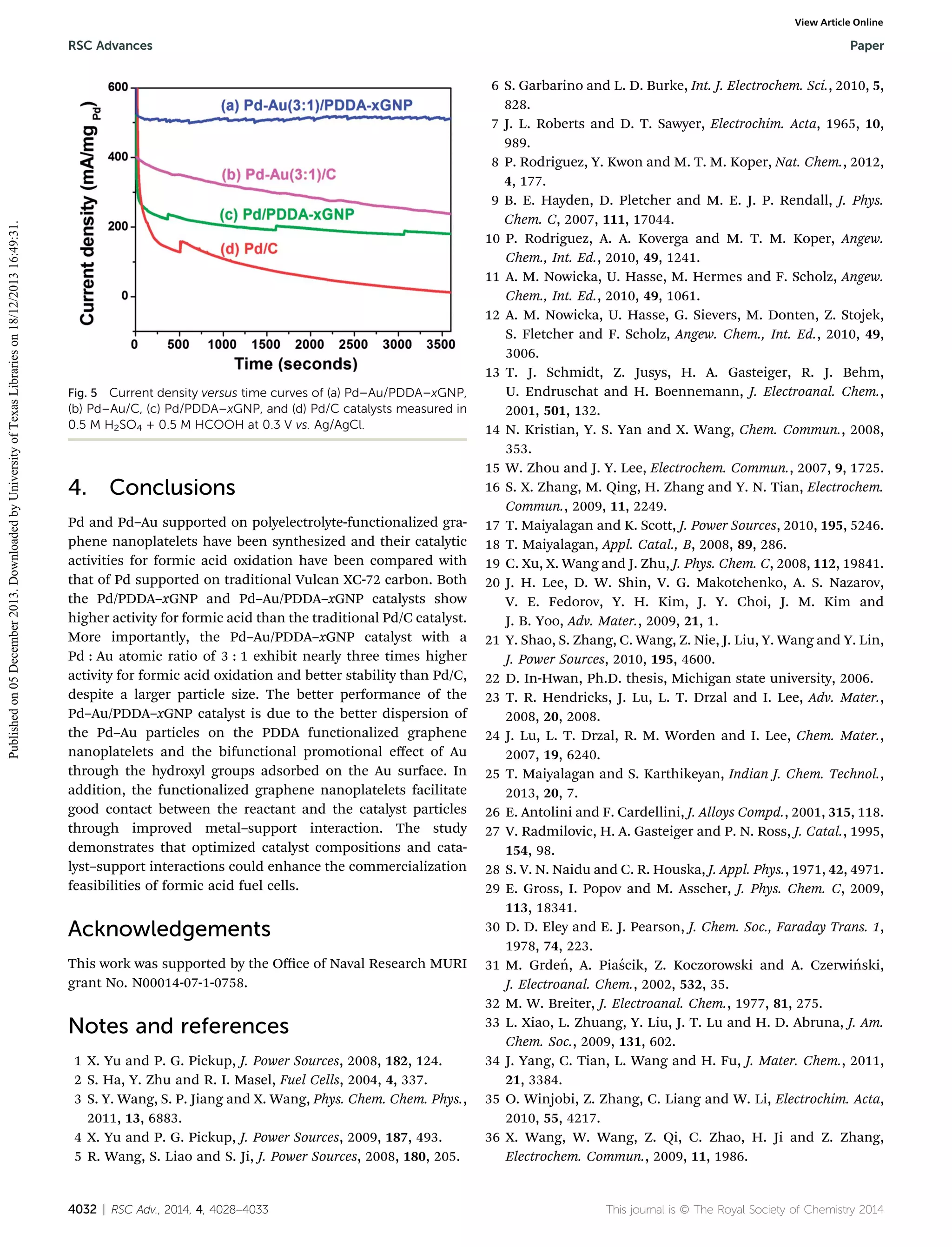


The study focuses on the synthesis and characterization of highly active palladium (Pd) and palladium-gold (Pd-Au) nanoparticles supported on functionalized graphene nanoplatelets for enhanced formic acid oxidation. Results indicate that the Pd-Au nanoparticles demonstrate superior catalytic activity for formic acid electro-oxidation compared to conventional Pd catalysts, attributed primarily to the alloying effect of Au, which mitigates catalyst poisoning. The research highlights the potential of these catalysts in direct formic acid fuel cells, emphasizing their advantages over traditional systems.


























































































