The document discusses clinical trials and good clinical practice (GCP) guidelines. It provides definitions and explanations of key concepts.
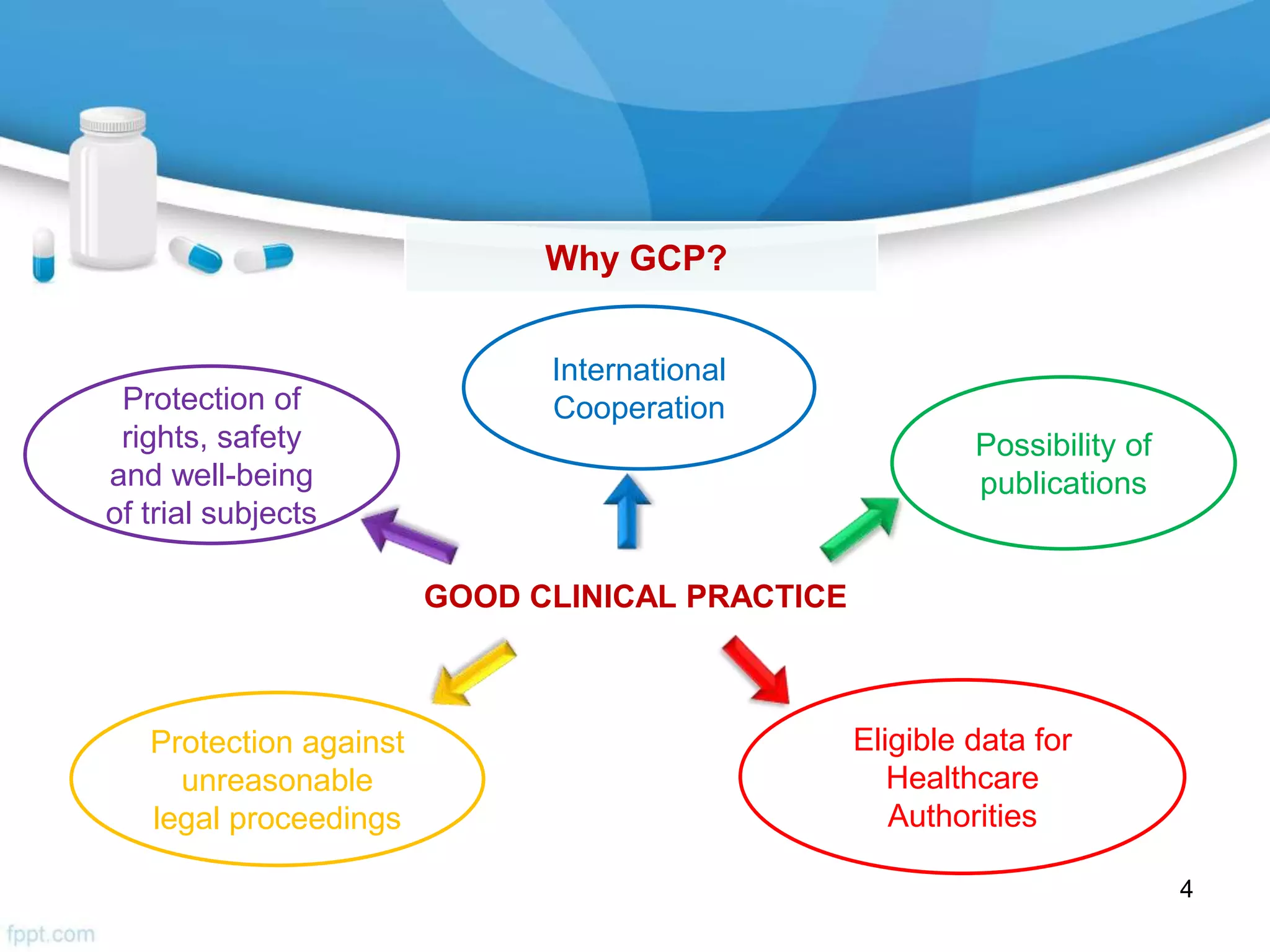
Specifically, it defines a clinical trial as an investigation in human subjects to discover or verify the effects of an investigational product. It describes GCP as international standards for clinical trial conduct that ensure data credibility and protect subject rights. The guidelines aim to harmonize standards across regions through organizations like the International Council for Harmonization.
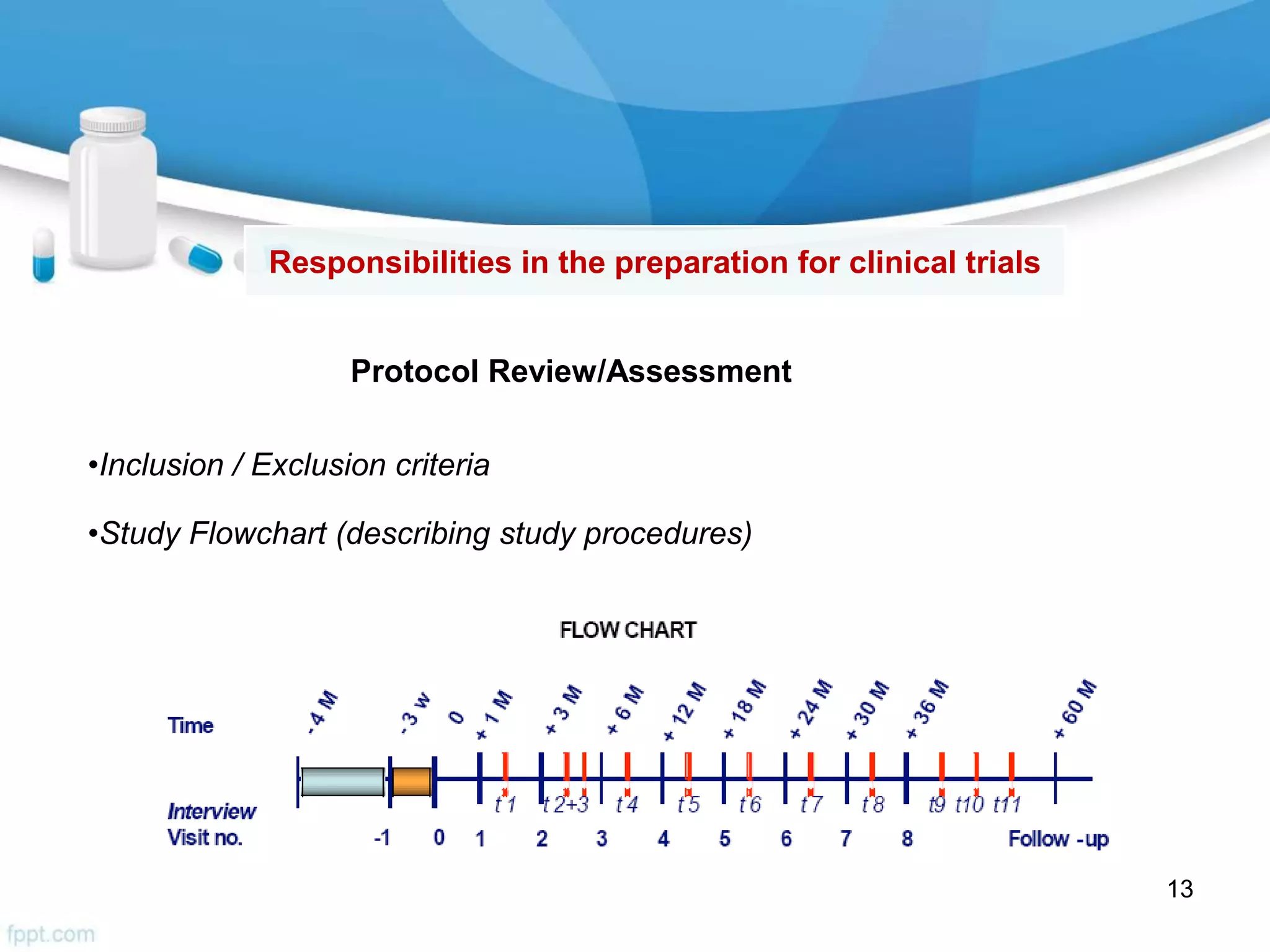
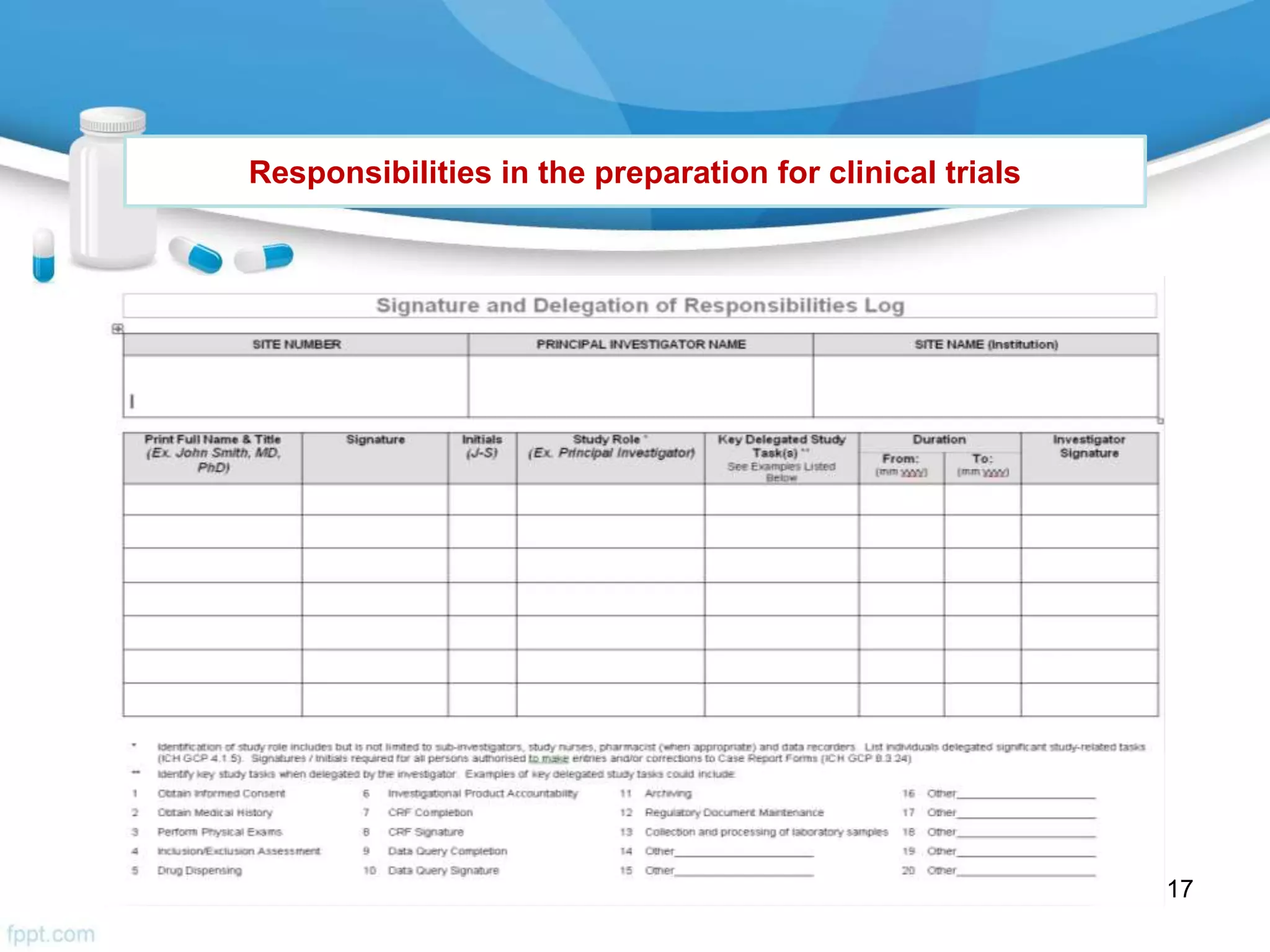
The document then outlines investigator responsibilities in areas like trial preparation, conduct, and closure to ensure compliance with GCP standards and protect subject safety and data integrity.