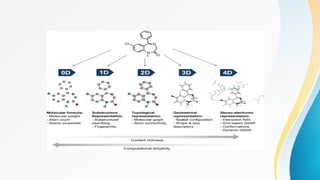
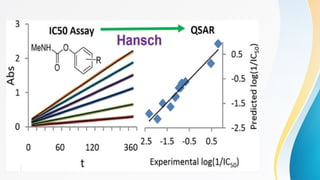
The document discusses the Quantitative Structure-Activity Relationship (QSAR), a method used to quantify the link between a drug's chemical structure and its biological activity. It outlines the historical development, types of QSAR models, and various physicochemical parameters such as hydrophobicity and steric factors that influence drug design. QSAR equations relate these properties to biological activity, aiding in the drug discovery process.