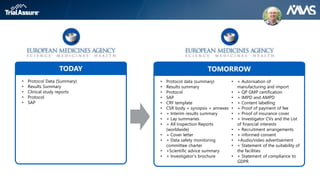
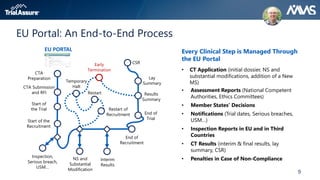
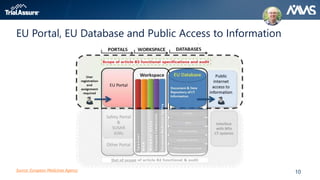
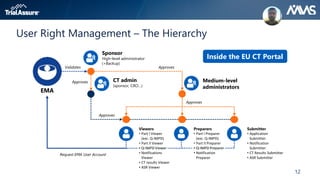
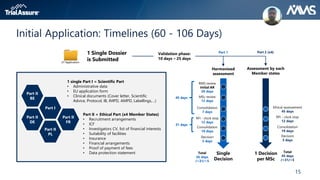
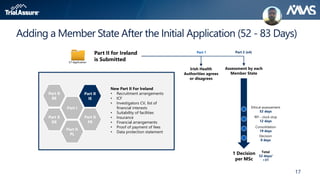
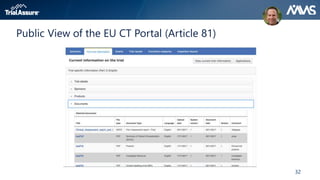
The webinar discusses key changes introduced by the new EU Clinical Trials Regulation that will revolutionize clinical trial transparency in Europe. Some of the major changes include a single application portal, expanded data disclosure requirements, and public access to clinical study documents and results. The new regulation aims to streamline the application process and increase oversight and transparency of clinical trials conducted in the European Union.























































![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)
























![CLINICAL_RESEARCH_REGULATIONS_IN_EUROPIAN_UNION_(_EMA[1] - Read-Only.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/clinicalresearchregulationsineuropianunionema1-read-only-240228102914-fed4af30-thumbnail.jpg?width=640&height=640&fit=bounds)

































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





