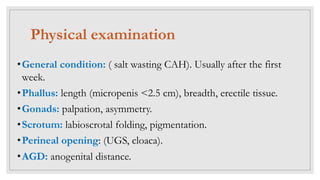
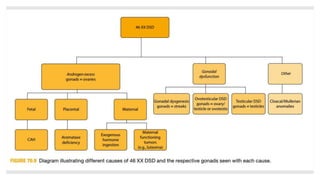
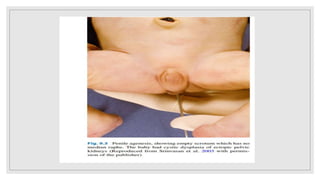
Case 1: A newborn boy with ambiguous genitalia including hypospadias, chordee, and undescended testes is found to have a 46,XY karyotype, indicating a disorder of androgen synthesis, action, or gonadal development. Further investigation including cystoscopy reveals a large prostatic utricle, which may require surgical correction after infancy.
Case 2: During surgery for an inguinal hernia in a 3-month-old girl, a testis is discovered in the hernia sac, indicating a potential disorder of sex development that requires further karyotyping and imaging to determine the diagnosis