Download to read offline












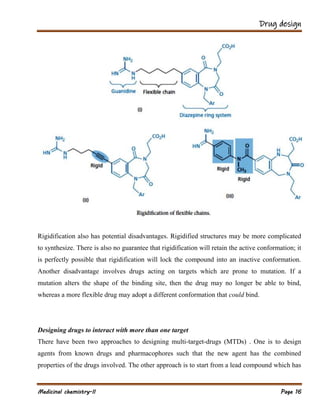


The document outlines various strategies in drug design and medicinal chemistry, emphasizing the importance of optimizing lead compounds through structural modifications to enhance efficacy and selectivity. It discusses techniques such as varying substituents, employing isosteres and bioisosteres, and rigidifying structures to improve binding interactions and reduce side effects. Additionally, the document addresses the design of multi-target drugs to combine the properties of existing drugs effectively.















































