Download as PDF, PPTX



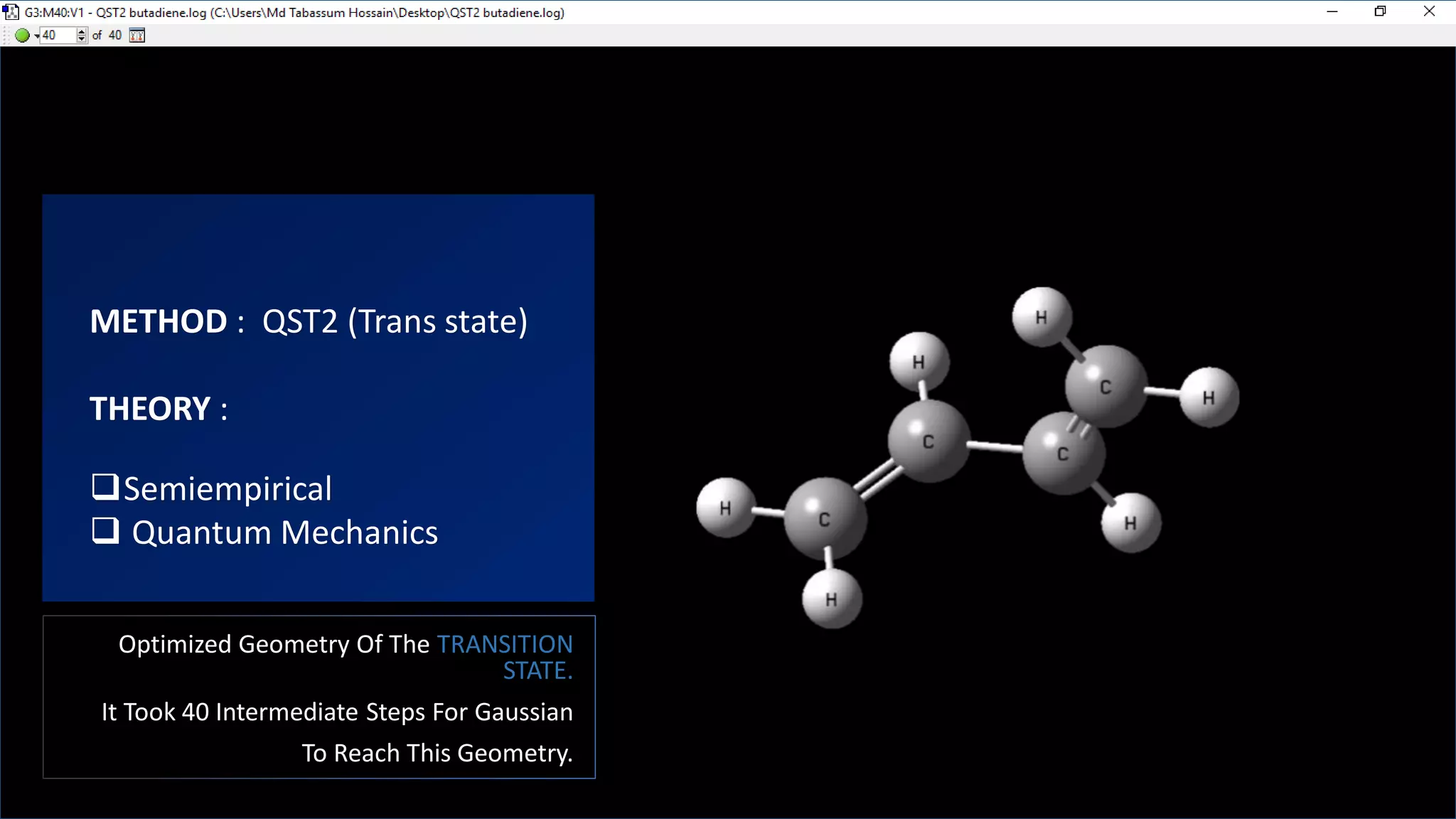


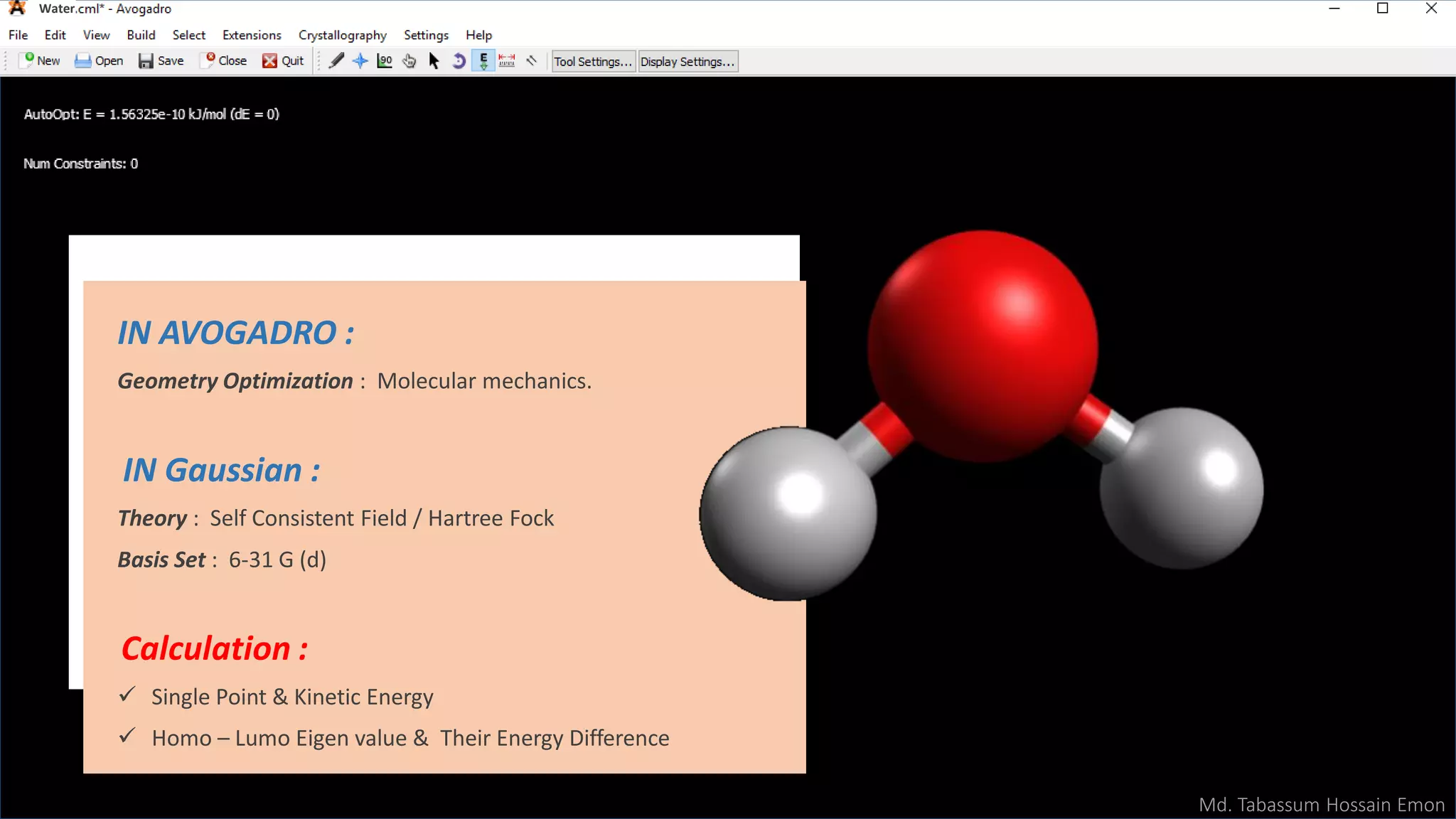










This document discusses computational chemistry calculations including HOMO-LUMO orbital energies, transition state geometry optimization, heat of combustion, and vibrational frequency calculations to generate IR spectra. Specific calculations mentioned include optimizing the transition state geometry of the reaction of 1,3-cis and 1,3-trans butadiene using Gaussian with 40 intermediate steps. HOMO and LUMO orbitals and energies are calculated for water using Gaussian with the 6-31G(d) basis set. Vibrational frequencies are calculated for paracetamol to generate an IR absorption spectrum graph.




















































