Downloaded 19 times



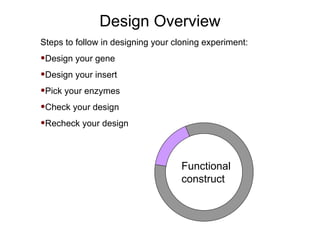
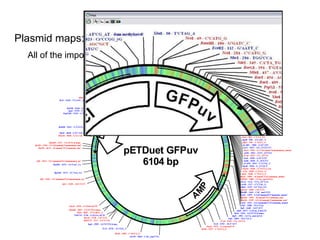


















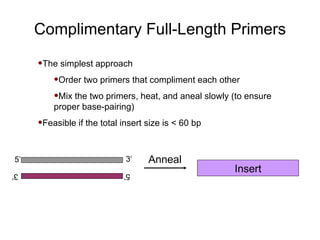











This document provides an overview of the cloning process and considerations for designing cloning experiments. It discusses four main steps: insert synthesis, restriction enzyme digestion, ligation, and transformation. Key aspects covered include gene and insert design using software like pDRAW32, choosing appropriate restriction sites and enzymes, primer design for insert synthesis, and vector and bacterial strain selection. The goal is to provide all the important information needed in one place to successfully clone a gene of interest.








































![Hemopoiesis[med]](https://cdn.slidesharecdn.com/ss_thumbnails/hemopoiesismed-110612232311-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



![Naturally acquired plasmodium knowlesi malaria in human, thailand[1]](https://cdn.slidesharecdn.com/ss_thumbnails/naturallyacquiredplasmodiumknowlesimalariainhumanthailand1-110301034416-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
































