Downloaded 420 times















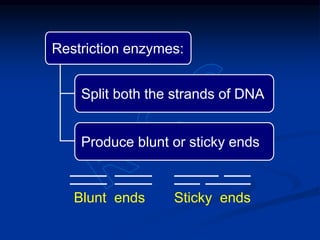



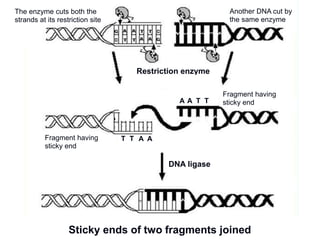




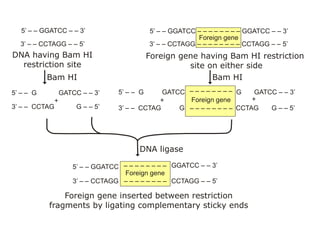








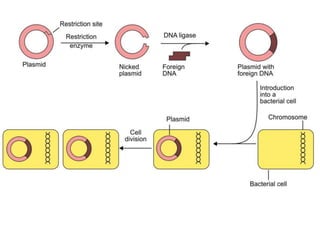






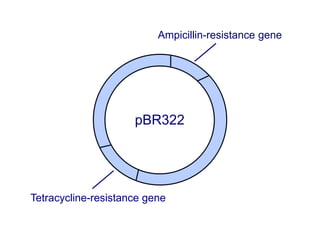
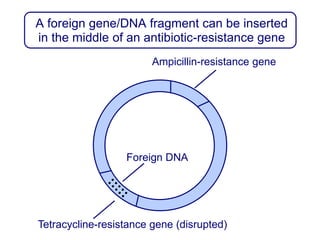



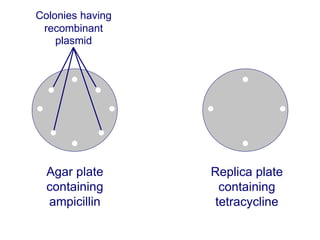

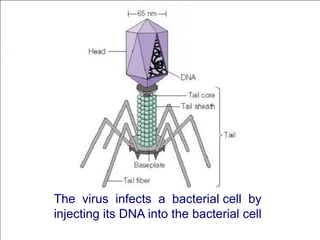
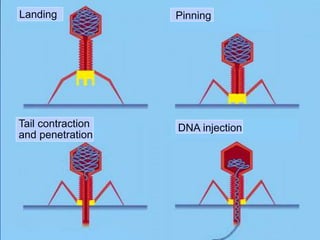
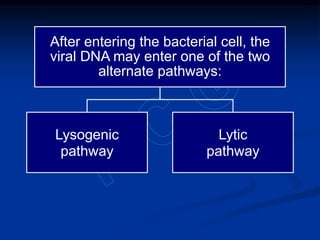






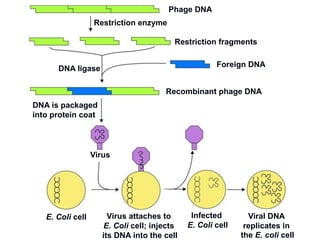



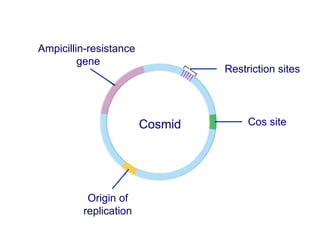


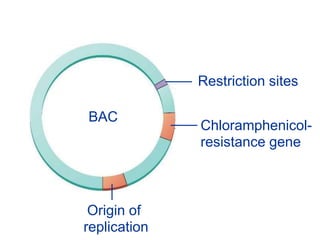




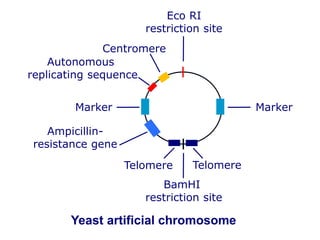










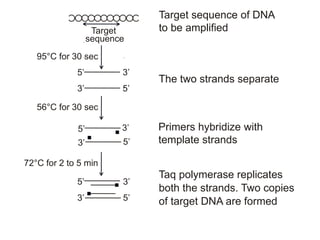
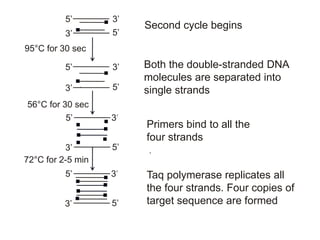






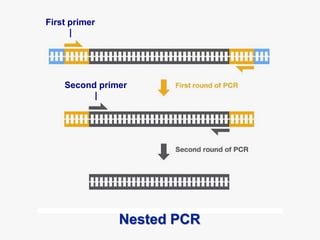

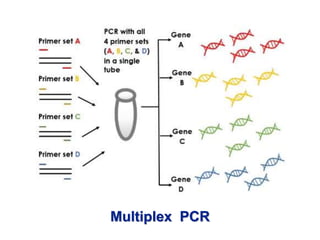







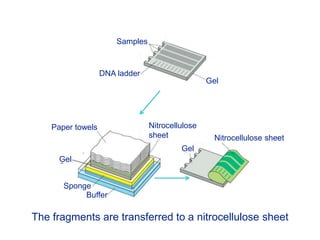

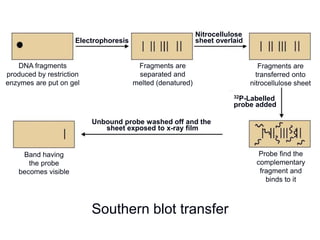

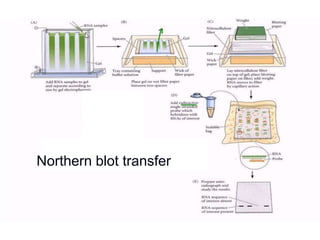

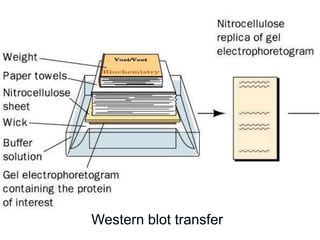
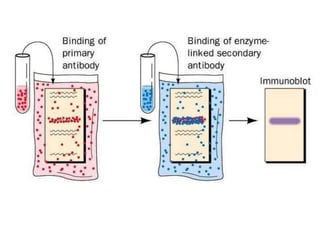








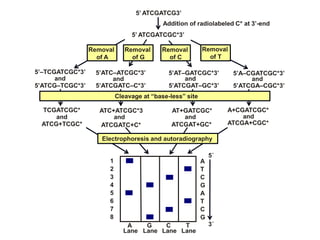

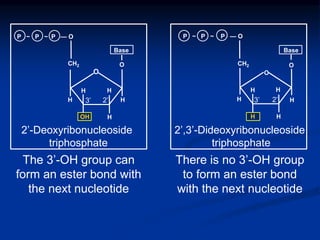










Recombinant DNA technology involves the combination of DNA fragments from different sources to create chimeric DNA, leading to significant advancements in various fields, including medicine and agriculture. Key tools include restriction endonucleases for DNA cleavage and ligases for joining DNA fragments, enabling the creation of recombinant plasmids and cloning vectors. Techniques such as polymerase chain reaction (PCR) allow rapid amplification of DNA, making it easier to study genes and develop applications like gene therapy and diagnostic tests.












































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)









