Downloaded 13 times


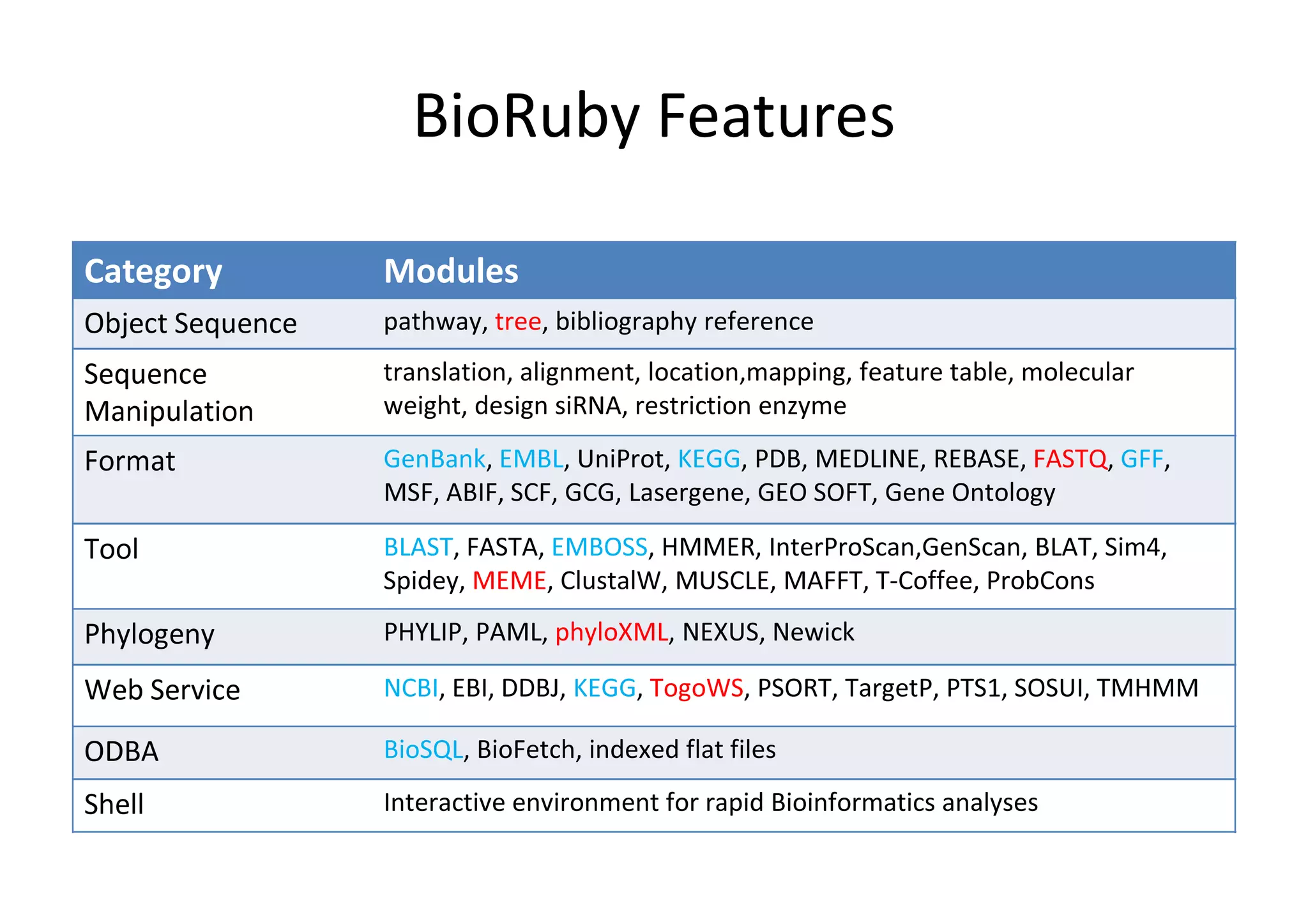

![Relevant New Features2
Bio::PhyloXML r/w by -Diana Jaunzeikare, Christian M Zmasek-
require ‘bio’ # libxml-ruby
#Create a parser
phyloxml = Bio::PhyloXML::Parser.new(‘example.xml’)
#Consume the tree
phyloxml.each do |tree|
puts tree.name
end
#Wrinting
writer = Bio::PhyloXML::Writer.new(‘my_tree.xml’)
write.writer(tree2)
#Extract information
phyloxml = Bio::PhyloXML::Parser.new(‘ncbi_taxnonomy_mollusca.xml’)
phyloxml.each do |tree|
tree.each_nome do |node|
print ‘Scientific name: ‘, node.taxonomies[0].scientific_name,‘n’
end
end Han, M. V. and Zmasek, C. M. (2009). phyloXML: XML for
evolutionary biology and
comparative genomics. BMC Bioinformatics, 10, 356.](https://image.slidesharecdn.com/bonnalbosc2010bioruby-100710215855-phpapp02/75/Bonnal-bosc2010-bio_ruby-9-2048.jpg)



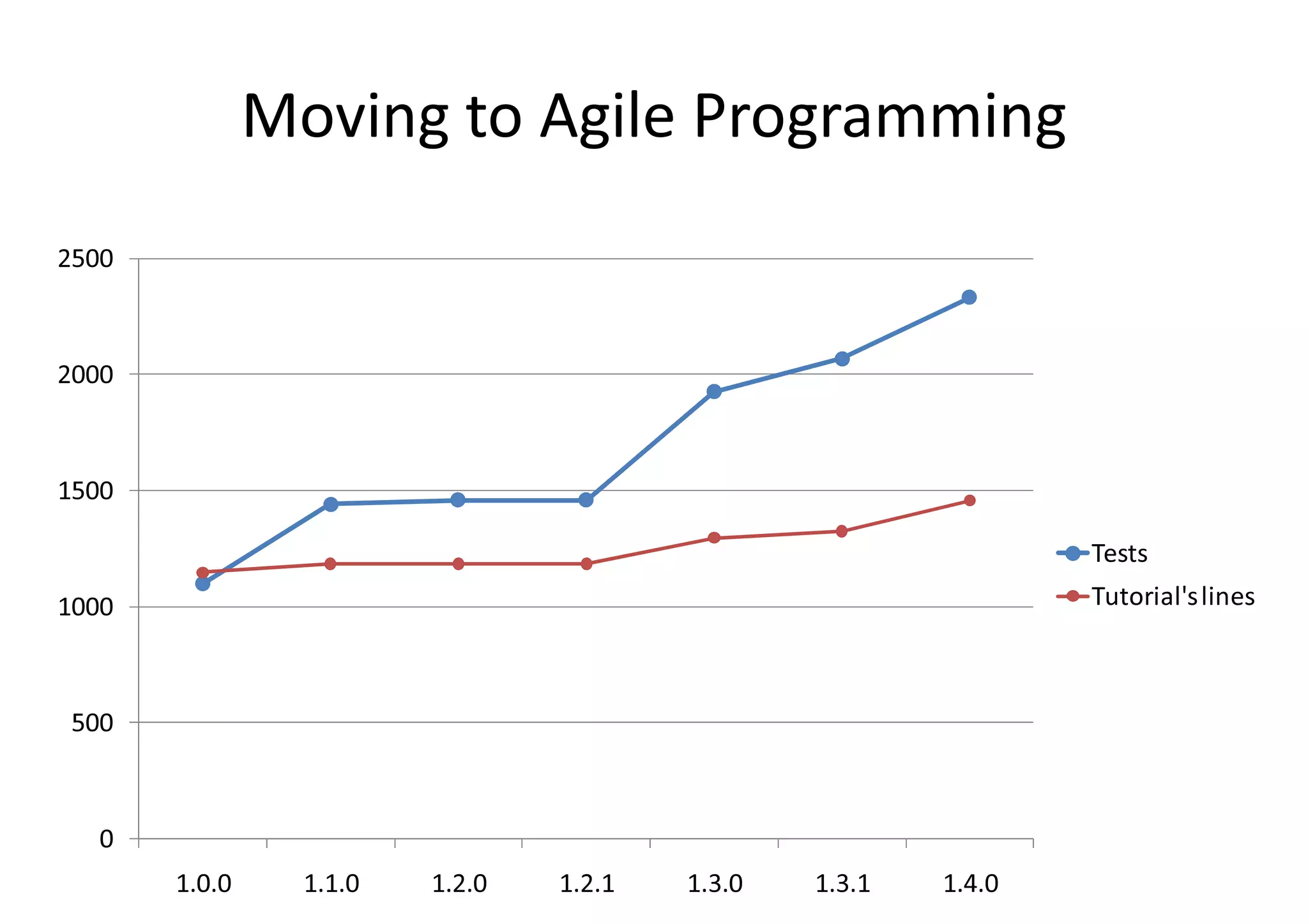
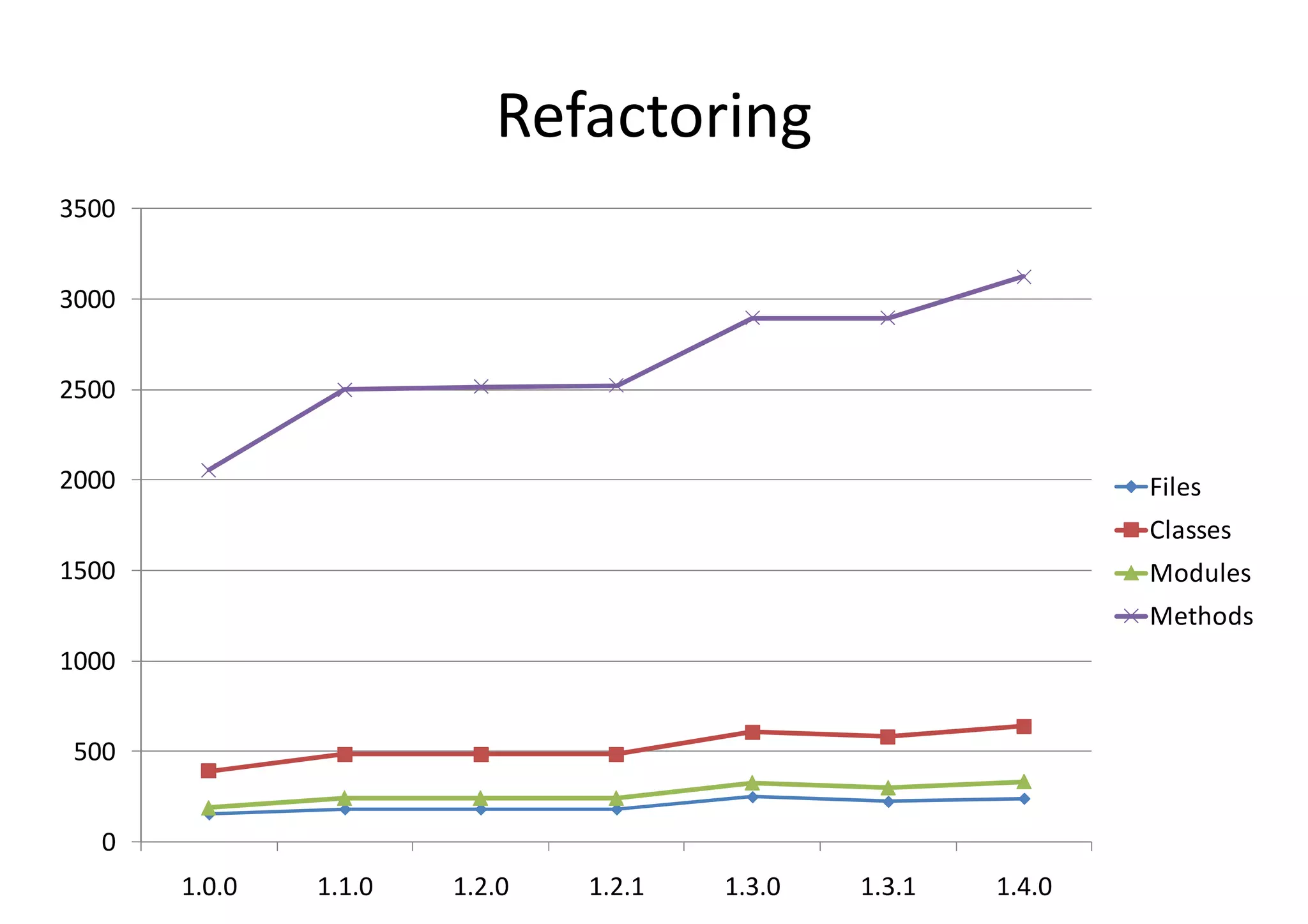







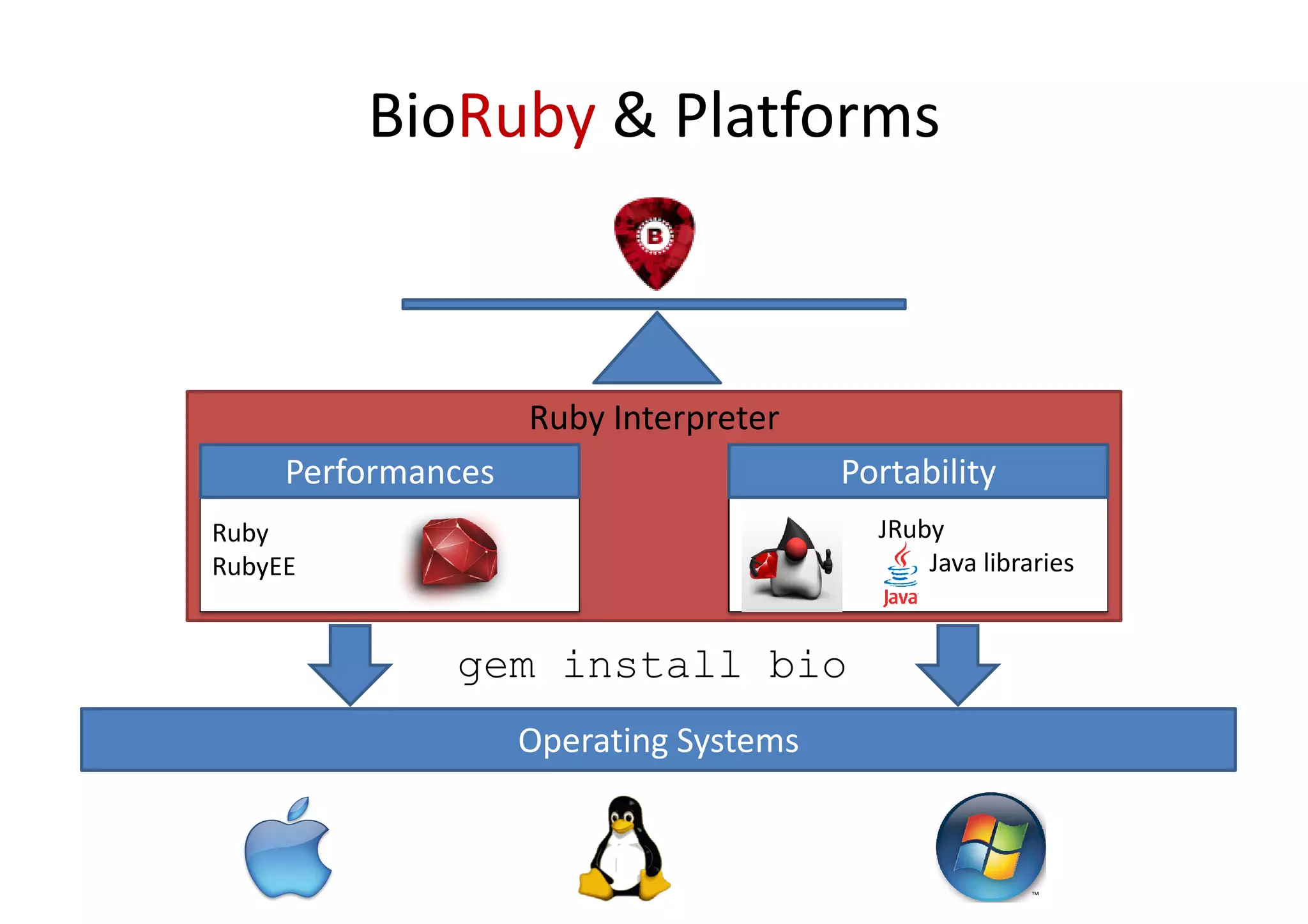
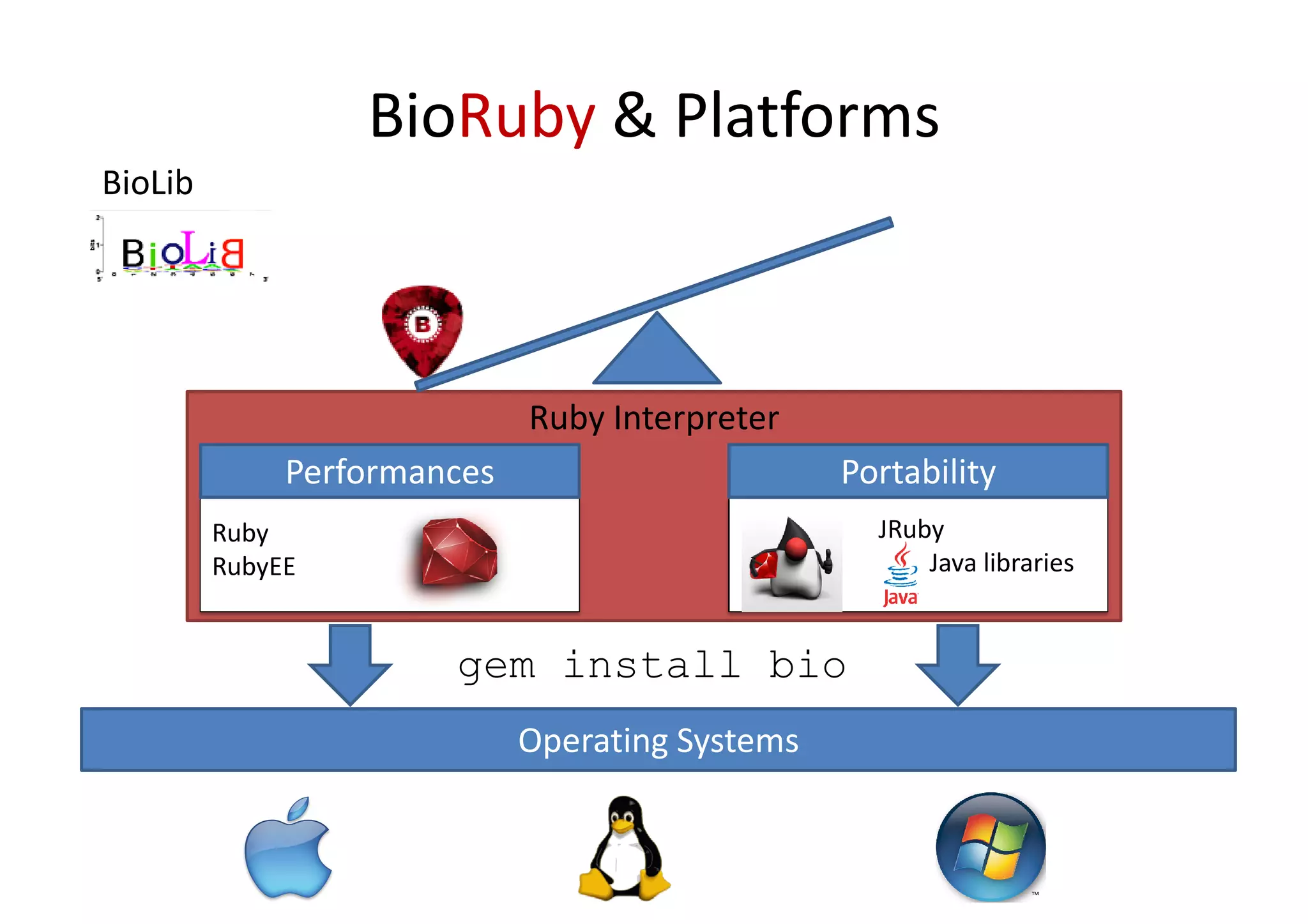
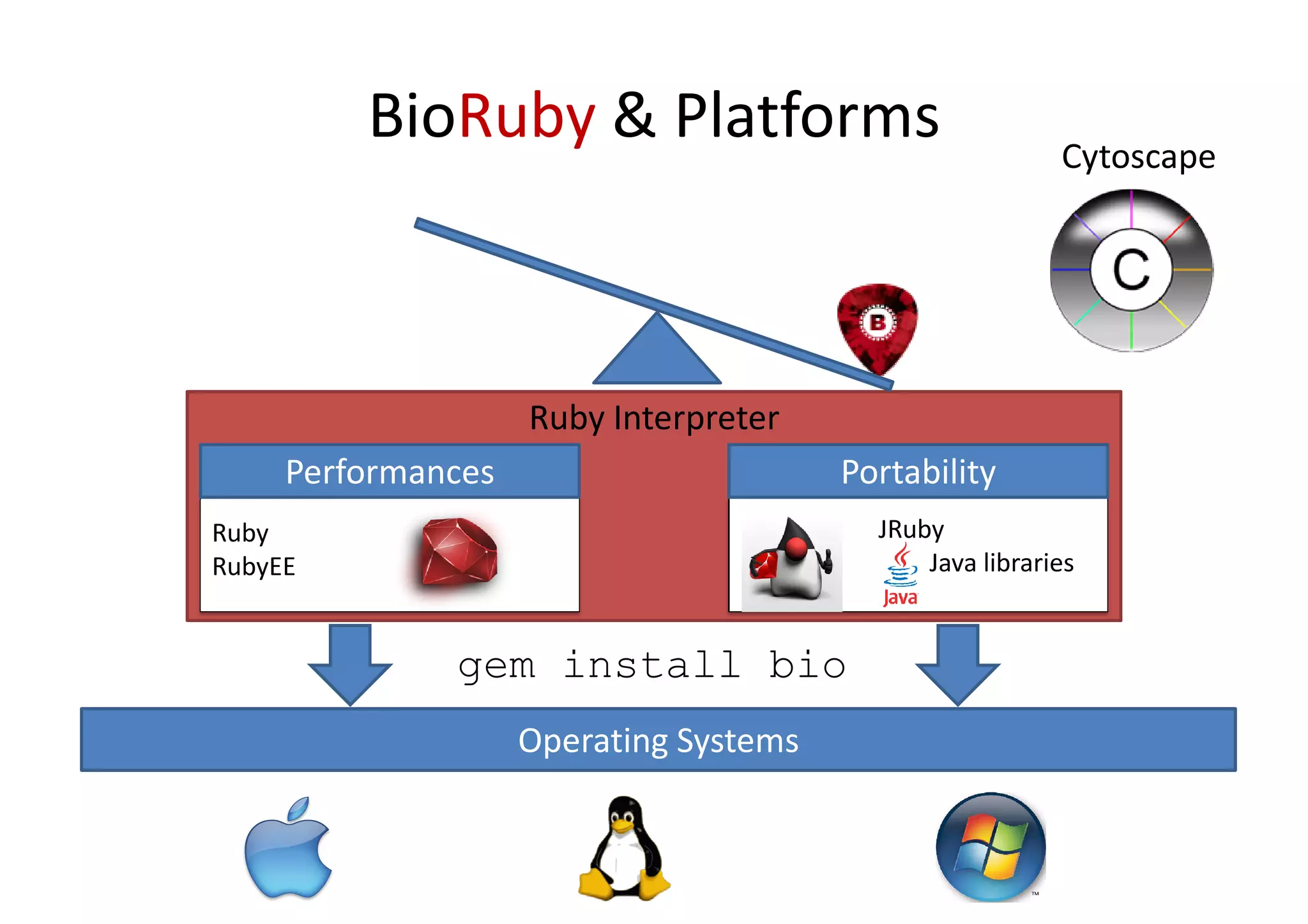
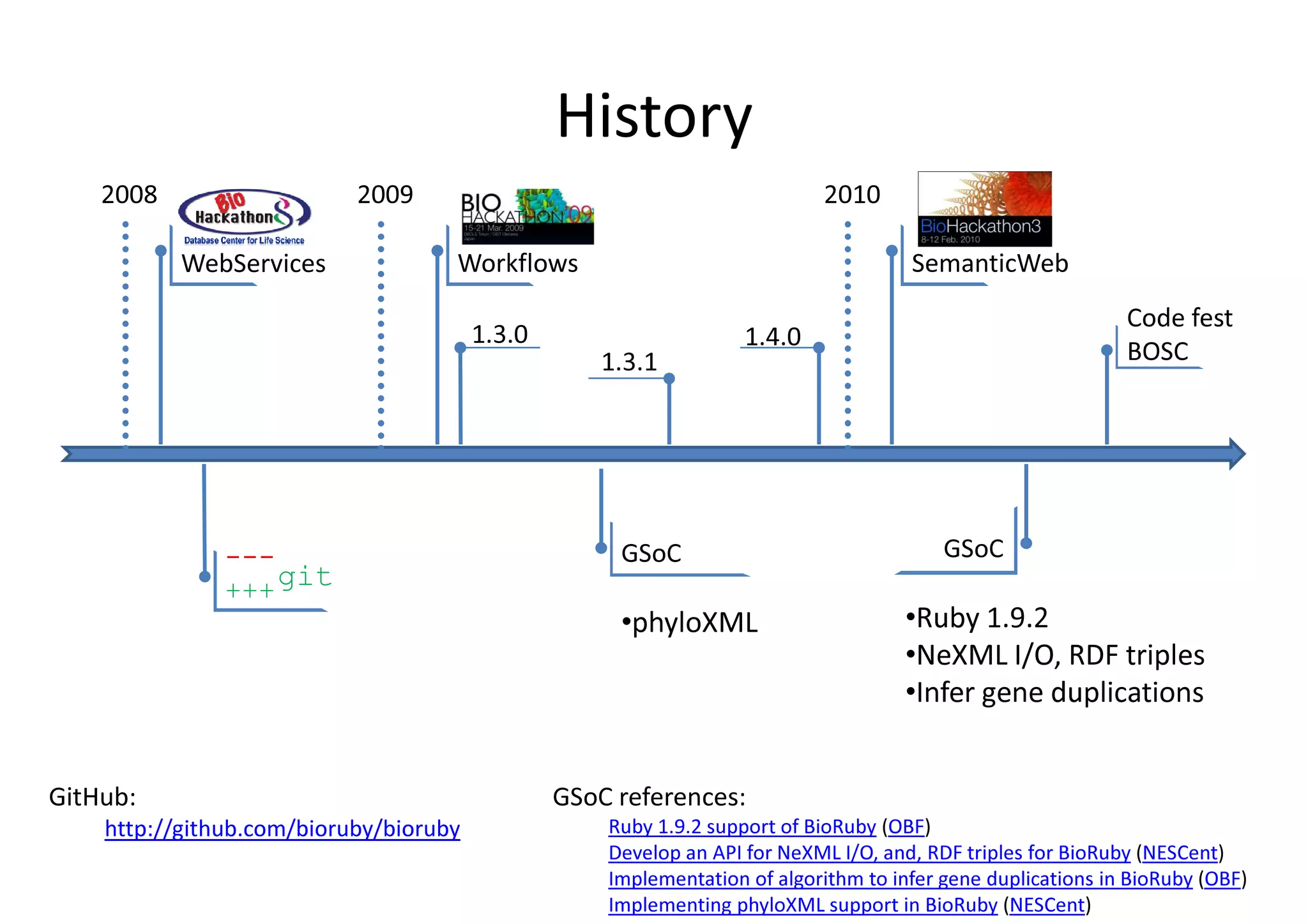
BioRuby is a bioinformatics library for the Ruby programming language. It provides object-oriented tools for tasks like sequence analysis, format conversion, running bioinformatics tools, and working with biological data. The latest version added features like improved support for phylogenetic XML (PhyloXML), next-generation sequencing FASTQ format reading/writing, and a REST API wrapper for the NCBI database. BioRuby development follows agile principles and its large developer community contributes new code frequently on GitHub. The project aims to improve integration with R and data visualization while maintaining a stable core.








































































![Vibe Coding vs. Spec-Driven Development [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/vibecodingvsspecdrivendevelopment-251209105622-43f455e7-thumbnail.jpg?width=640&height=640&fit=bounds)












