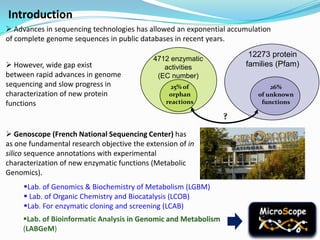
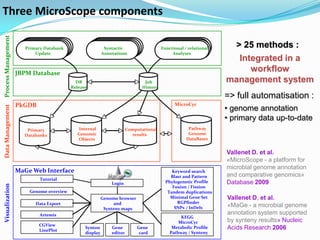
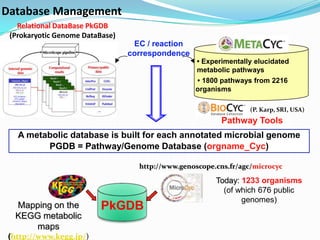
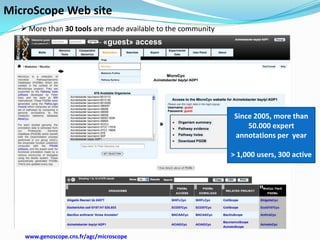
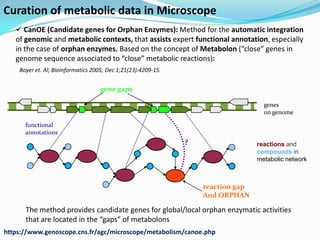
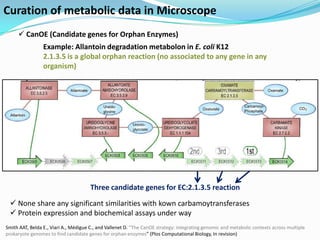
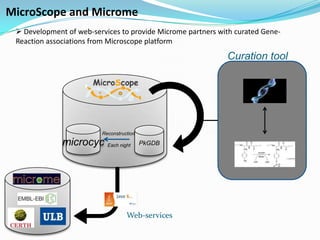
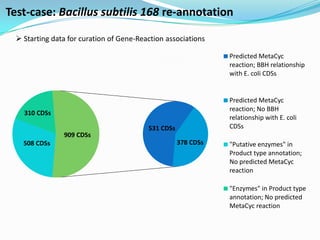
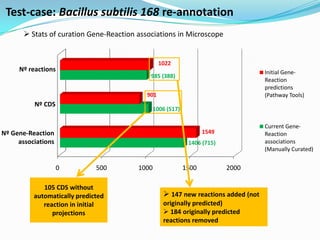
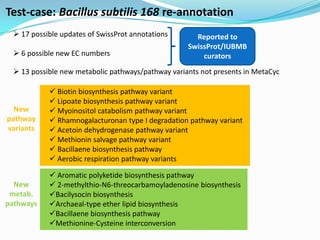
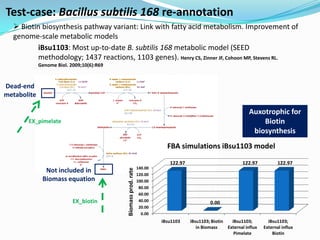
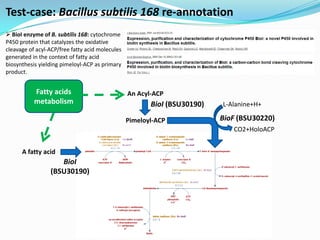
The document outlines the efforts of the LabGem team at Genoscope to enhance genomic and metabolic annotations using bioinformatics tools and methodologies. It highlights the gaps in the characterization of orphan proteins and enzymes, the role of various laboratories in extending metabolic genomics, and the development of systems for integrating data to assist in enzyme function discovery. The Canoe strategy and the curation of metabolic data, such as gene-reaction associations, are emphasized as pivotal in mapping and validating metabolic pathways across numerous microbial genomes.























![Consortium to produce_bio_fuels_from_jatropha[1]](https://cdn.slidesharecdn.com/ss_thumbnails/consortiumtoproducebiofuelsfromjatropha1-100816001937-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




















































![Getting Started with Apache Spark: Big Data Made Simple [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/apachesparkgettingstarted-260203175547-8361bcc3-thumbnail.jpg?width=640&height=640&fit=bounds)


