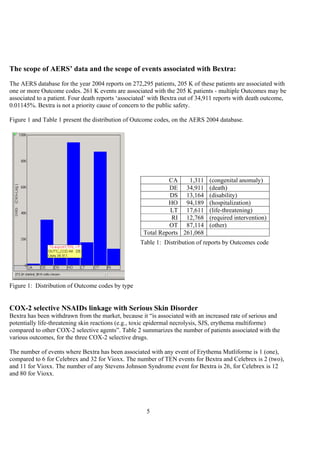
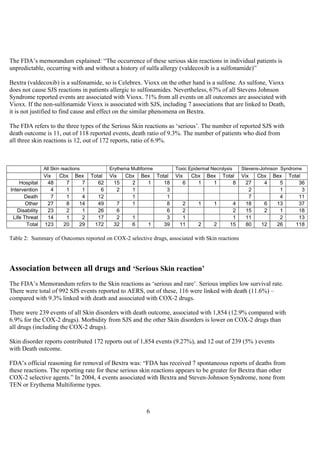
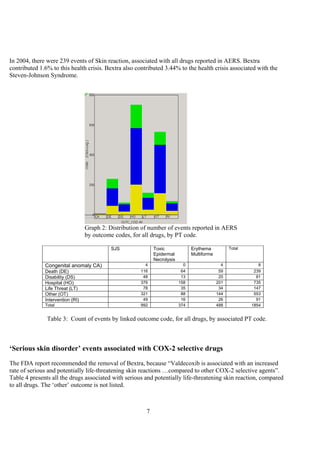
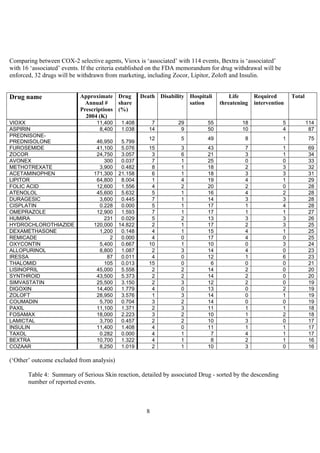
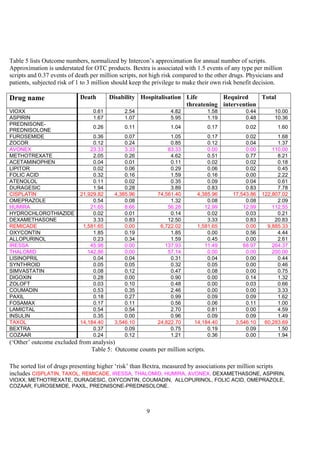
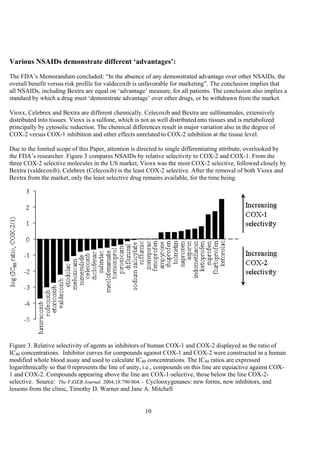
The document discusses the FDA's April 2005 decision to request that Pfizer voluntarily withdraw Bextra from the market or face formal withdrawal procedures. It summarizes the FDA's analysis from a memorandum that cited an increased rate of serious skin reactions with Bextra compared to other NSAIDs. However, the document argues that the AERS data used by the FDA to make its decision has significant limitations and was not designed for determining actual drug risks. It claims the FDA's standards could require withdrawing many other drugs and questions whether the decision to withdraw Bextra was justified. The document recommends Bextra and Vioxx be reinstated and that the FDA re-evaluate how it assesses drug risks and makes major regulatory decisions.








































































![Relationship Between Pharmacovigilance And Patient Safety[1]](https://cdn.slidesharecdn.com/ss_thumbnails/relationshipbetweenpharmacovigilanceandpatientsafety1-13120005672929-phpapp02-110729233628-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)