Downloaded 73 times




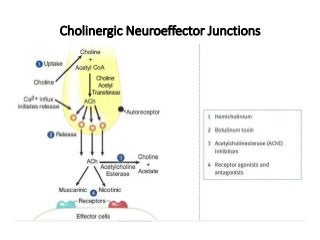
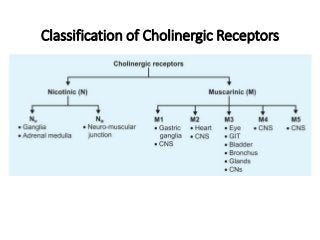
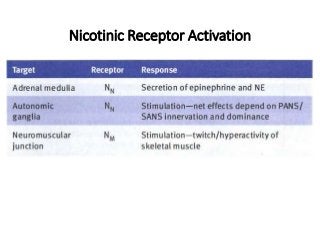
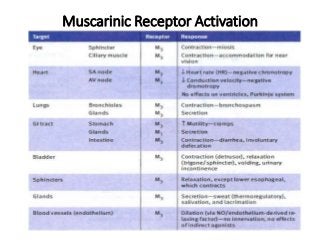
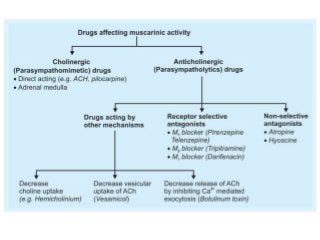









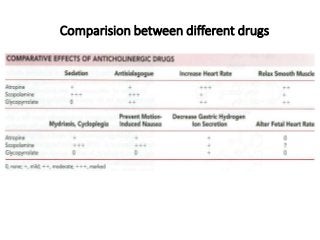




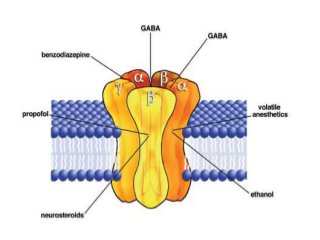
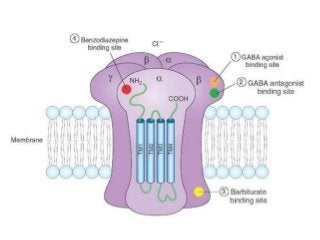


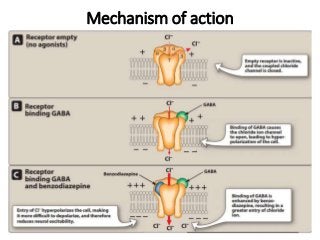
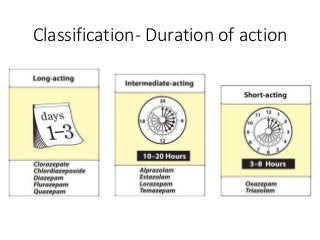











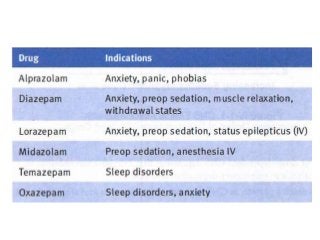
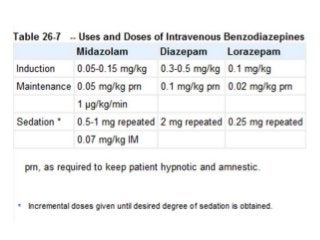


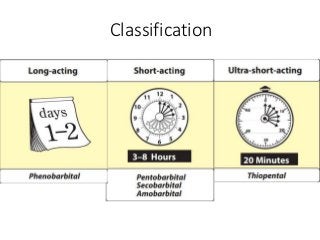
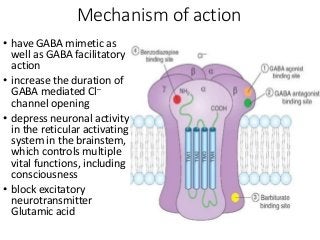





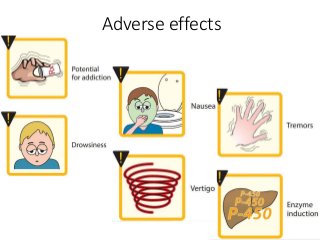
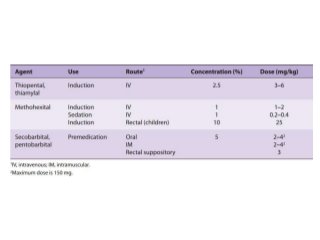





The document discusses the roles and uses of anticholinergics and sedatives in anesthetic practice, focusing on the autonomic nervous system and the effects of various medications. It covers the mechanisms, pharmacokinetics, and pharmacodynamics of anticholinergic drugs, such as atropine and scopolamine, as well as benzodiazepines, barbiturates, and alpha-2 agonists, emphasizing their uses, side effects, and interactions. The content targets medical professionals looking to understand the applications of these drugs in anesthesia and sedation management.























































































