Download as PDF, PPTX











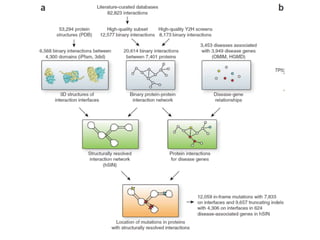
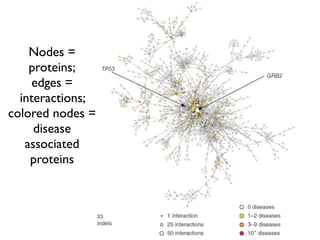

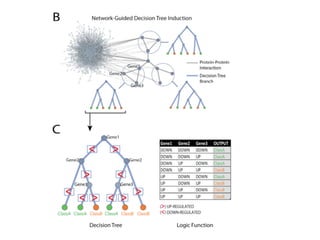
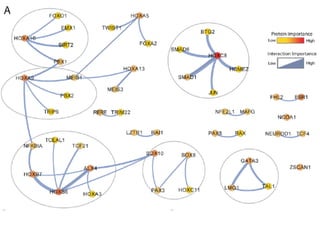

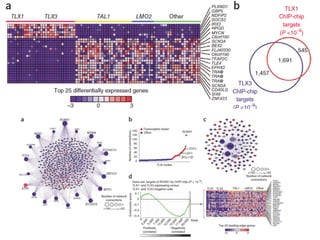

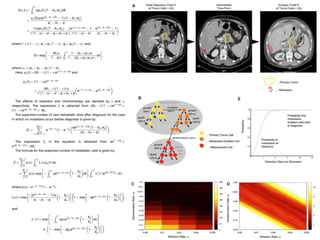
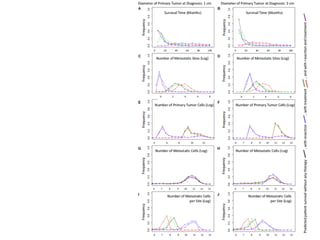
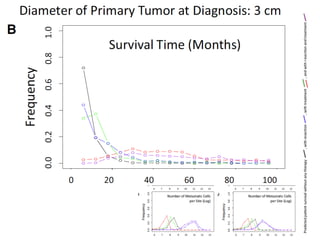

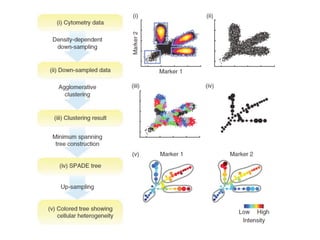
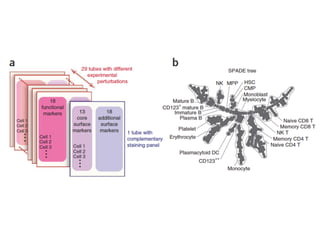

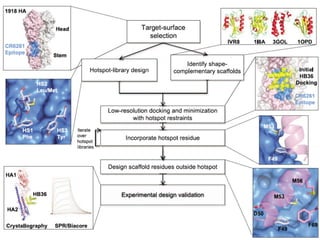
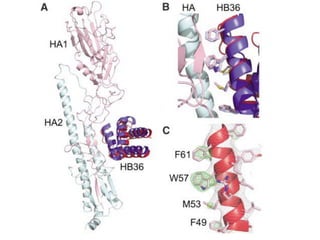





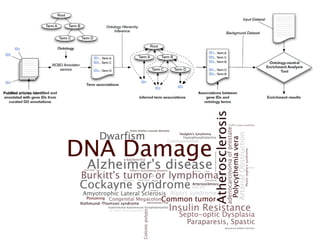

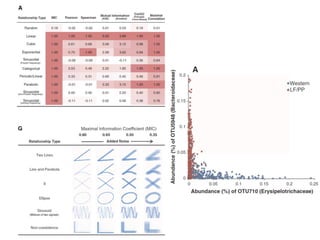

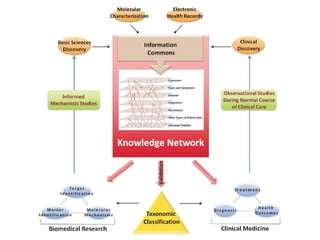



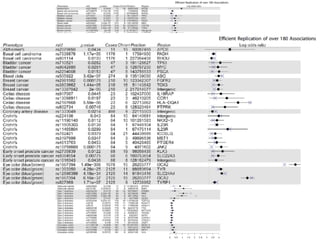

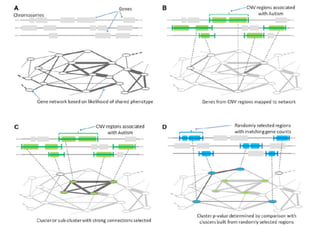
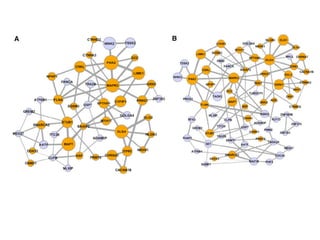
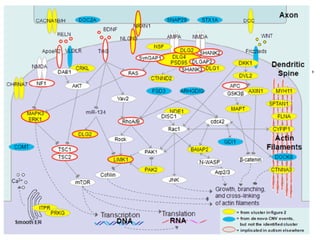


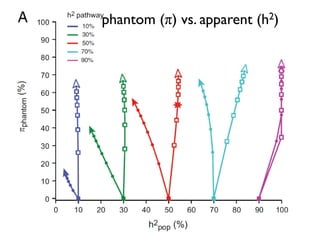

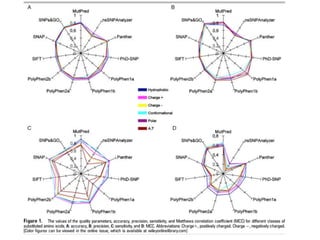

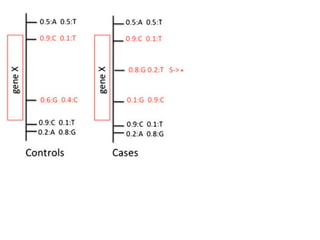
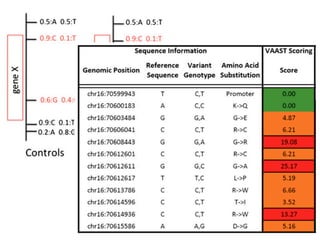

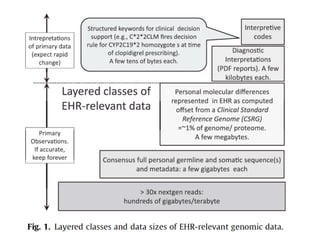



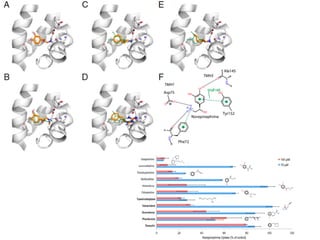

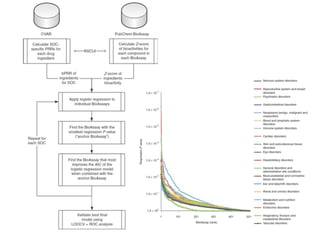

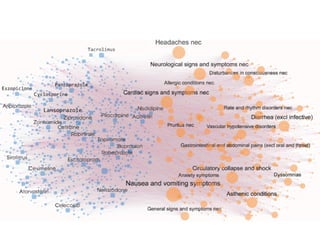


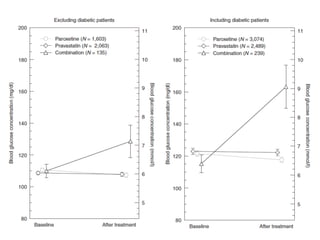


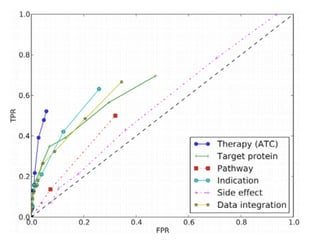
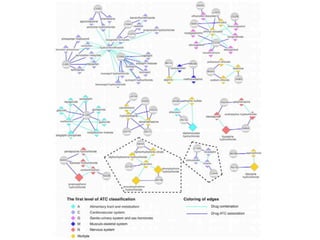


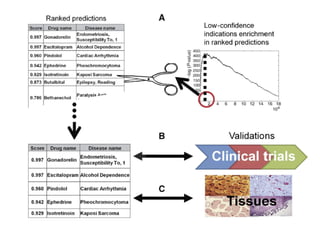

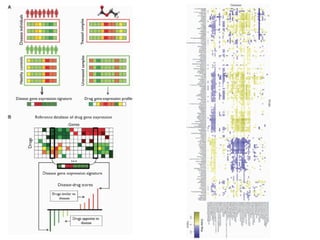







This document provides a summary of the 2012 Translational Bioinformatics conference. It highlights several important papers presented at the conference in areas like systems medicine, finding and defining phenotypes, biomarkers, and genomic infrastructure. The document outlines the goals of the conference, the process used to select papers, caveats about the selection, and thanks various contributors. It then briefly summarizes several key papers from the conference in these areas.









































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)







