This document outlines a pediatric epilepsy slide deck presented by the American Epilepsy Society. It covers several key topics:
Section 1 discusses seizures and epilepsy syndromes that present in neonates and early infancy, including Ohtahara syndrome, early myoclonic encephalopathy, benign familial neonatal epilepsy, and others.
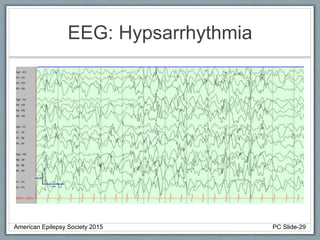
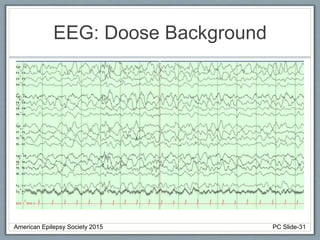
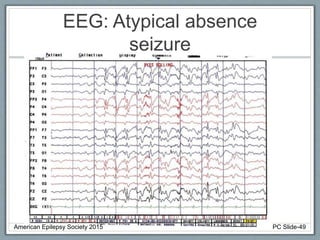
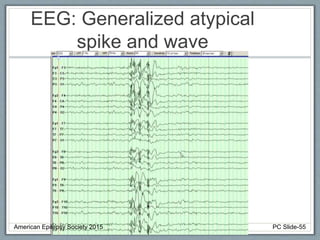
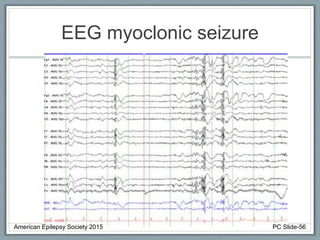
Section 2 covers epilepsy syndromes that present in early childhood and adolescence, such as West syndrome (characterized by epileptic spasms and a hypsarrhythmia EEG pattern), Doose syndrome, and Lennox-Gastaut syndrome.
Section 3 describes unique etiologies of epilepsy that often present with pediatric onset. Section 4 discusses surgical evaluation of int


























































![Alper’s hepatopathic
poliodystrophy
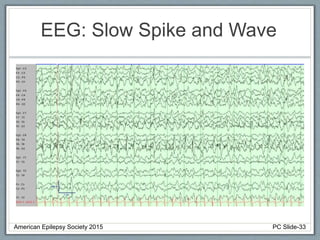
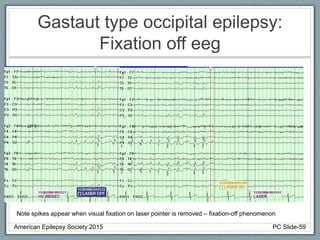
• EEG: Slow or absent posterior dominant rhythm with
multifocal and generalized epileptiform activity.
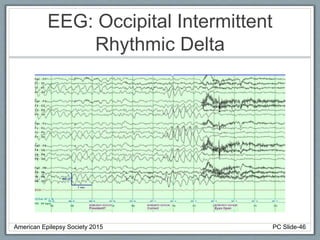
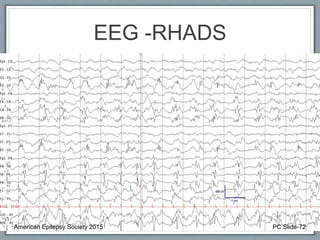
Rhythmic High Amplitude Delta with Superimposed
Spikes [RHADS] common associated EEG pattern.
(see figure)
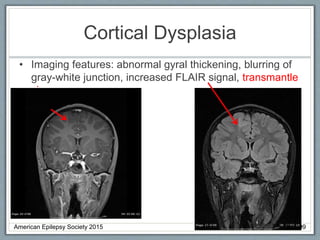
• Imaging: Variable, including migratory cortical and
subcortical hyperintensities, basal ganglia and thalami
changes, diffuse white matter changes, cerebellar
atrophy
• Clinical course: progressively worsening epilepsy and
hepatic failure. Death within months to 12 years of
onset.
American Epilepsy Society 2015 PC Slide-71](https://image.slidesharecdn.com/aespedworkgroupslidefinal-240307040912-6aae7367/85/AES-Pediatrics-Working-group-Slide-Final-pptx-71-320.jpg)













































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)




![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)













