Download as PDF, PPTX
















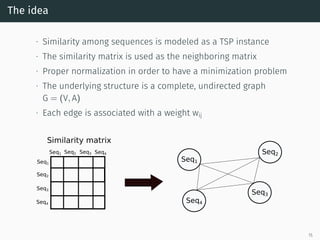






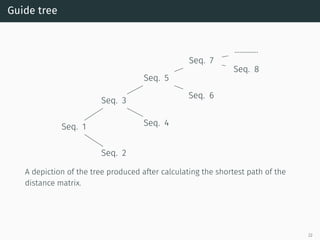


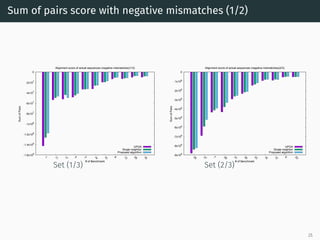
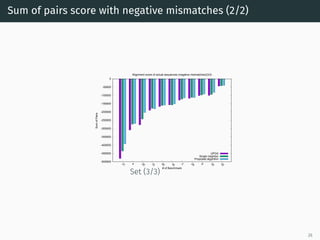
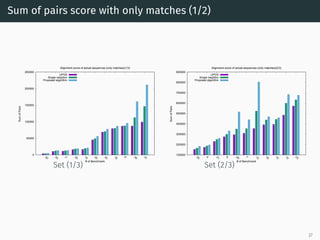
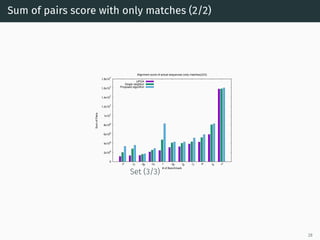







The document presents a quantum-inspired heuristic for solving the multiple sequence alignment problem in bioinformatics. It models the sequence similarity as a traveling salesman problem instance using a normalized similarity matrix. The method applies a quantum-inspired generalized variable neighborhood search metaheuristic to approximate the shortest Hamiltonian path and generate an initial alignment. Evaluation on real biological sequences shows it outperforms progressive methods, producing alignments with good sum-of-pairs scores, especially for large sequence sets.





















































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)