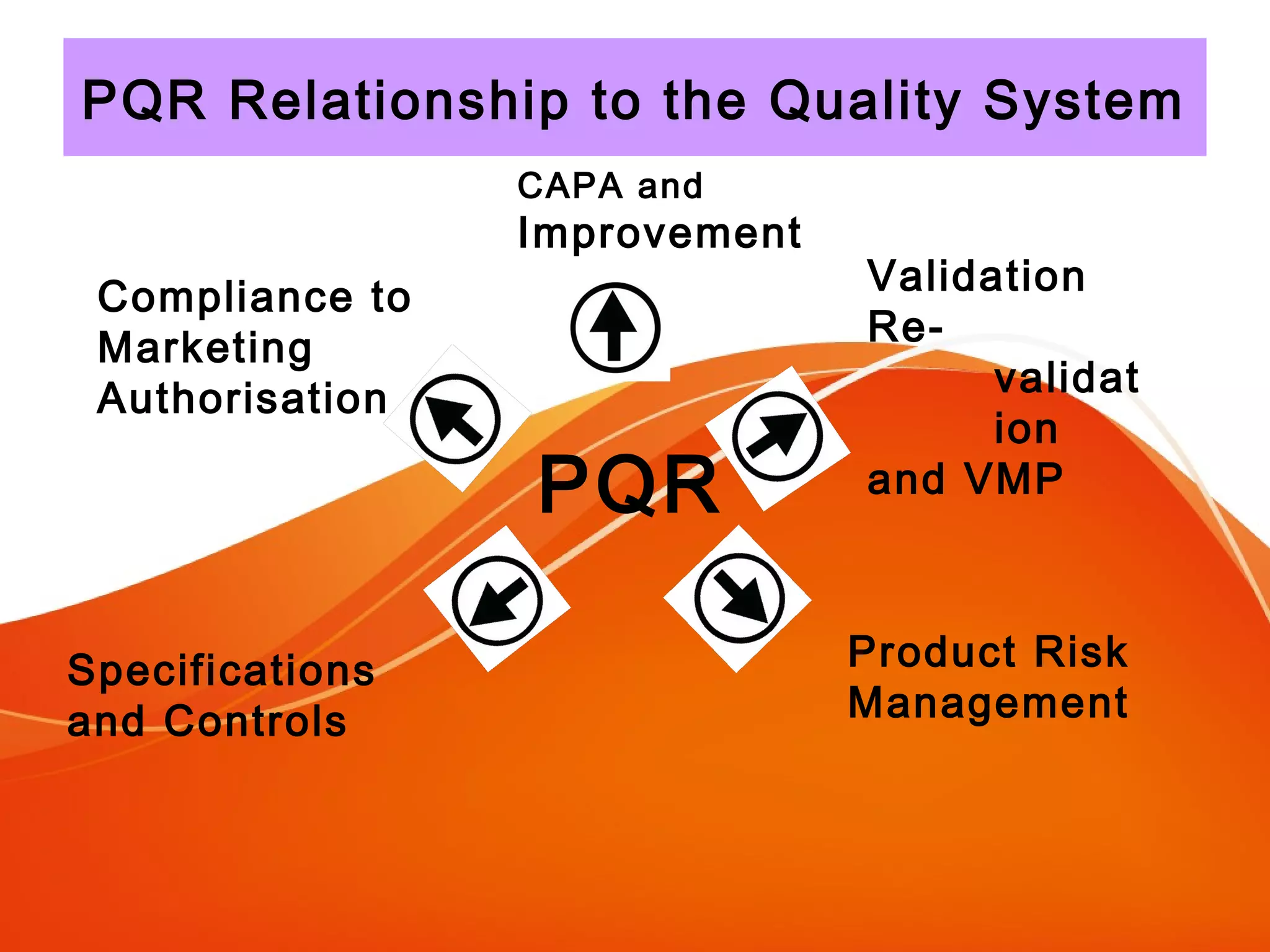
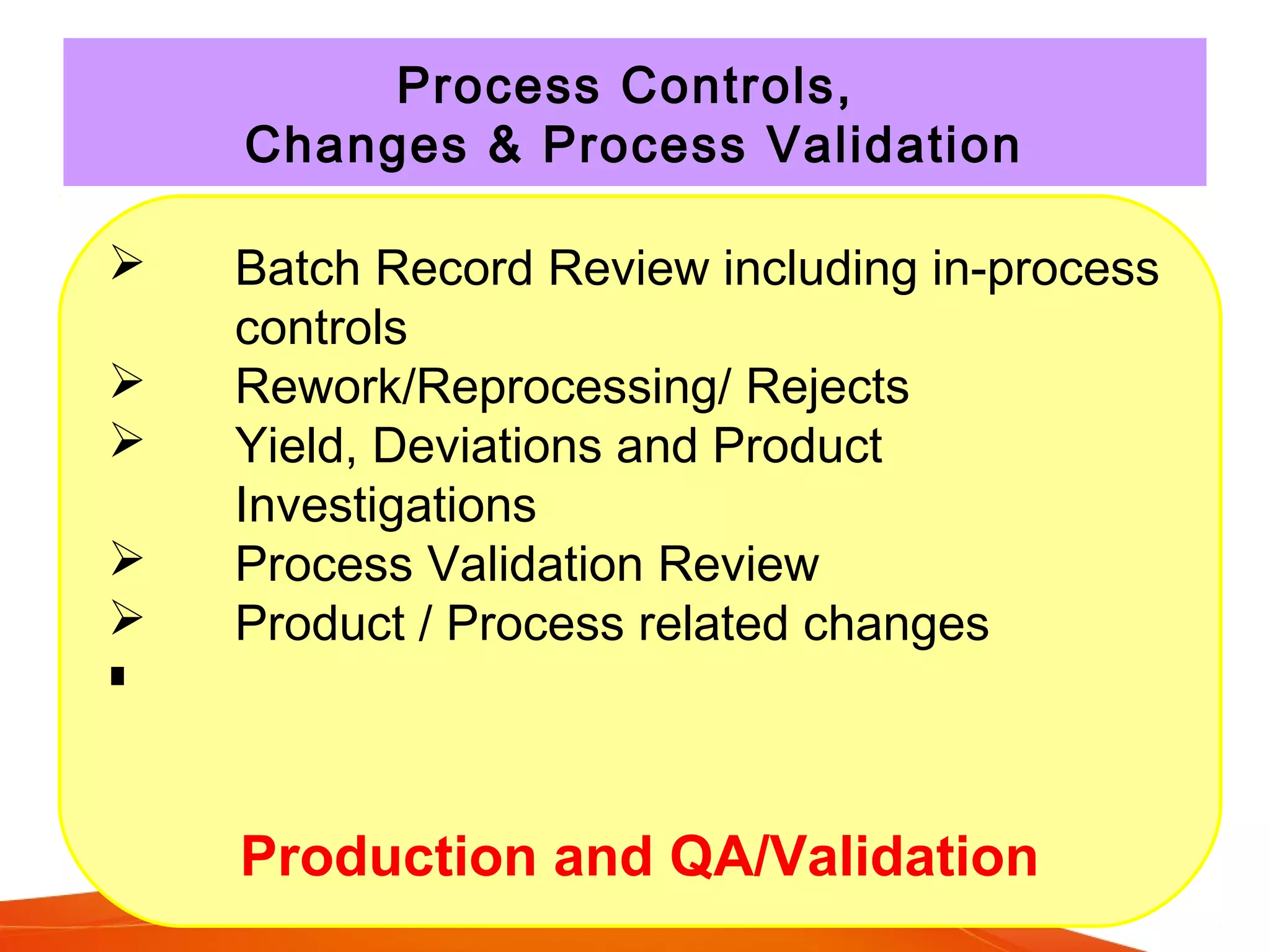
The Product Quality Review (PQR) is a regular review of all licensed medicinal products conducted to verify consistency of manufacturing processes and the appropriateness of specifications. The objectives of the PQR include determining the need for process, specification or validation changes; verifying compliance; identifying trends; and determining corrective actions. The EU requires annual PQRs that review areas like starting materials, process and product testing results, failed batches, deviations, changes made, and stability monitoring results. The PQR is intended to enhance quality and identify improvements.