Downloaded 1,589 times














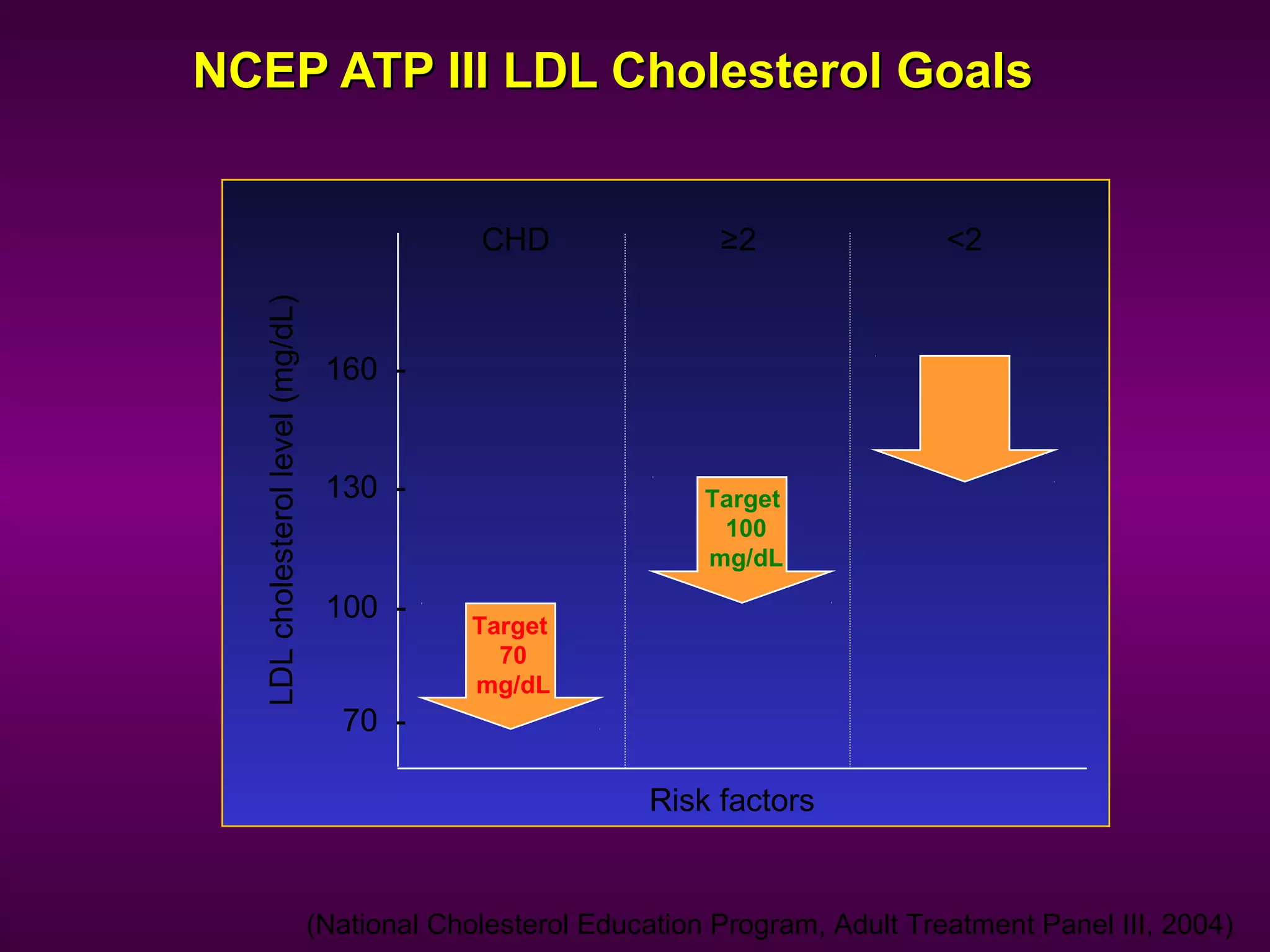
















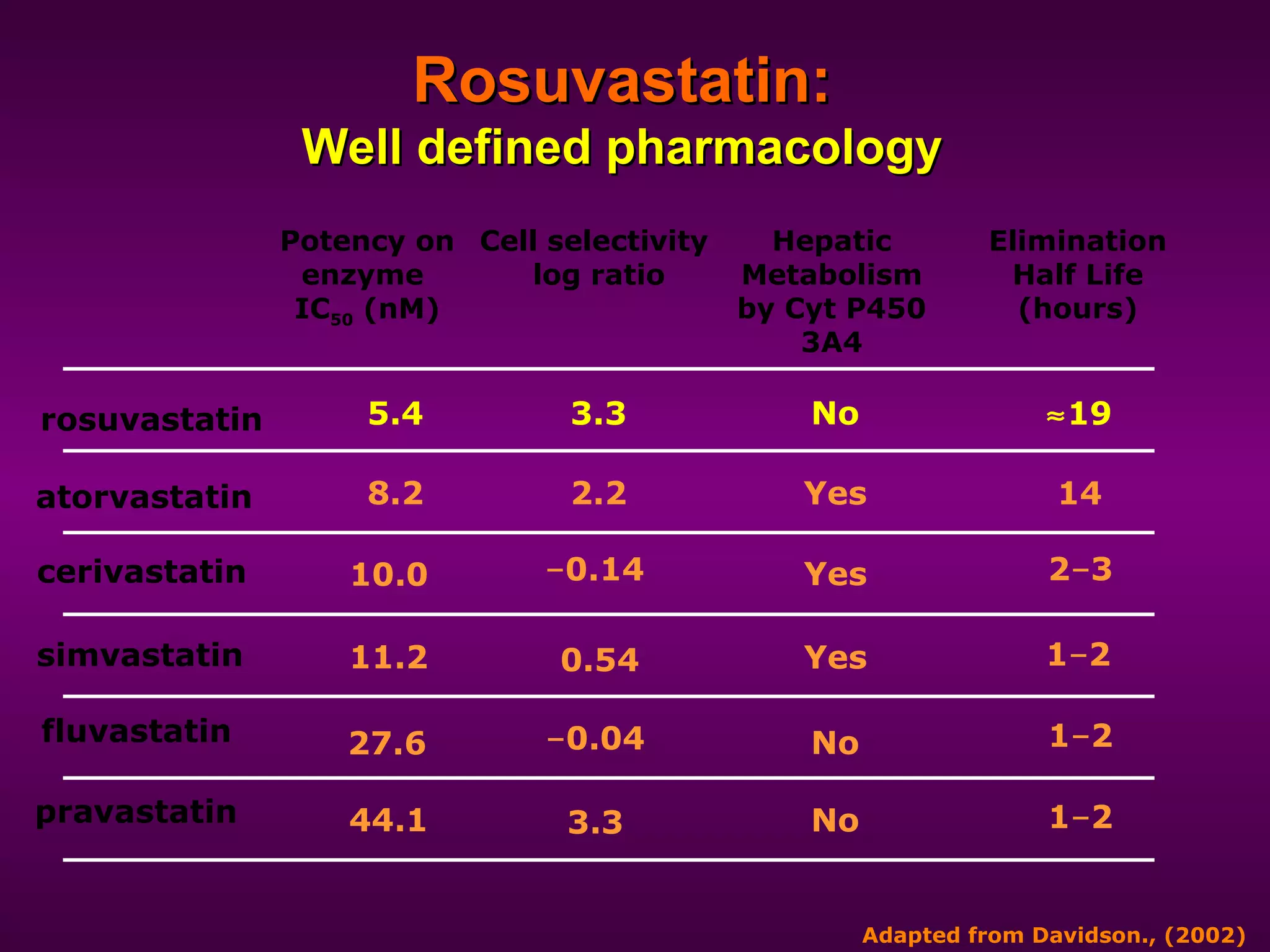





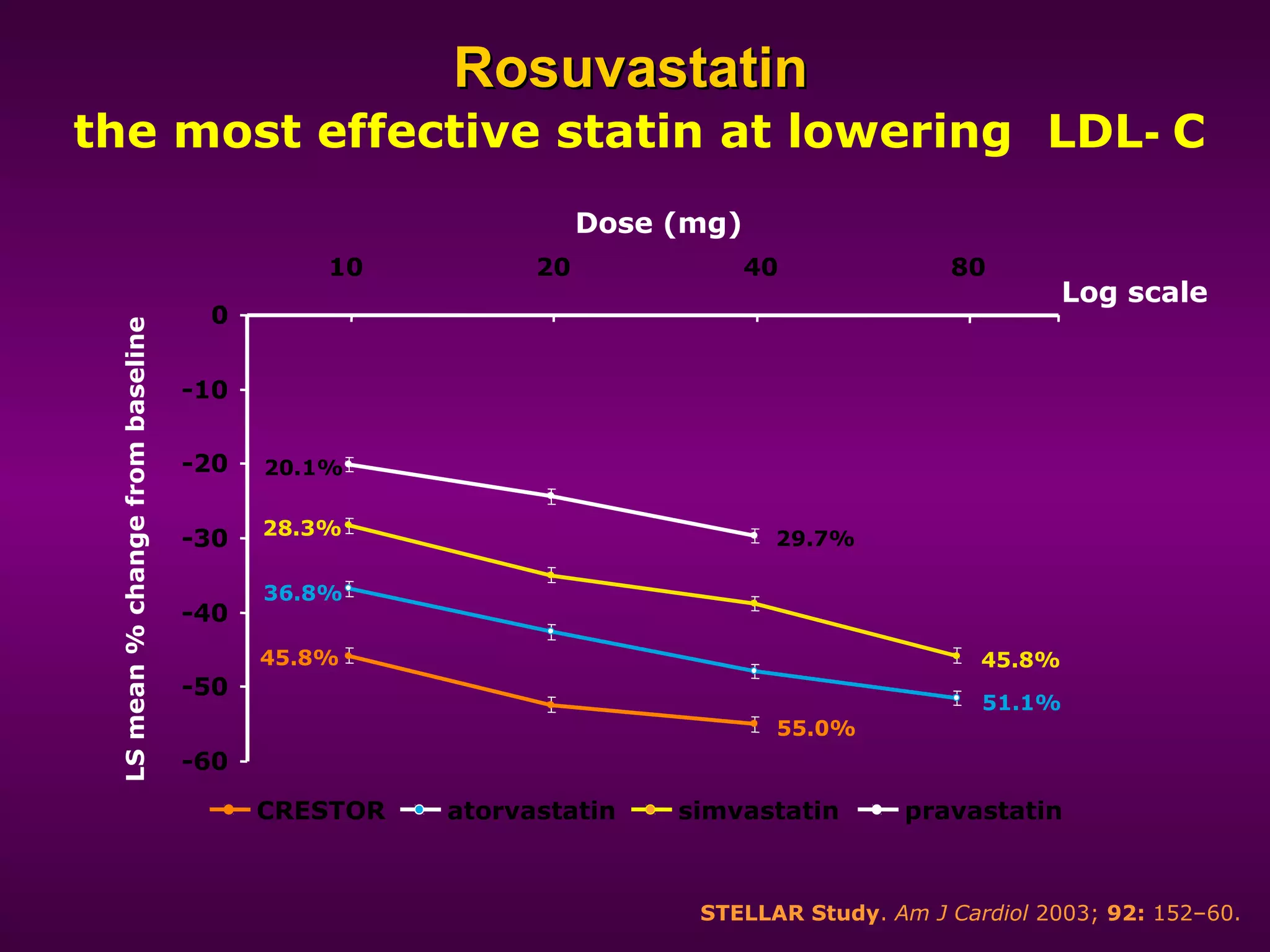


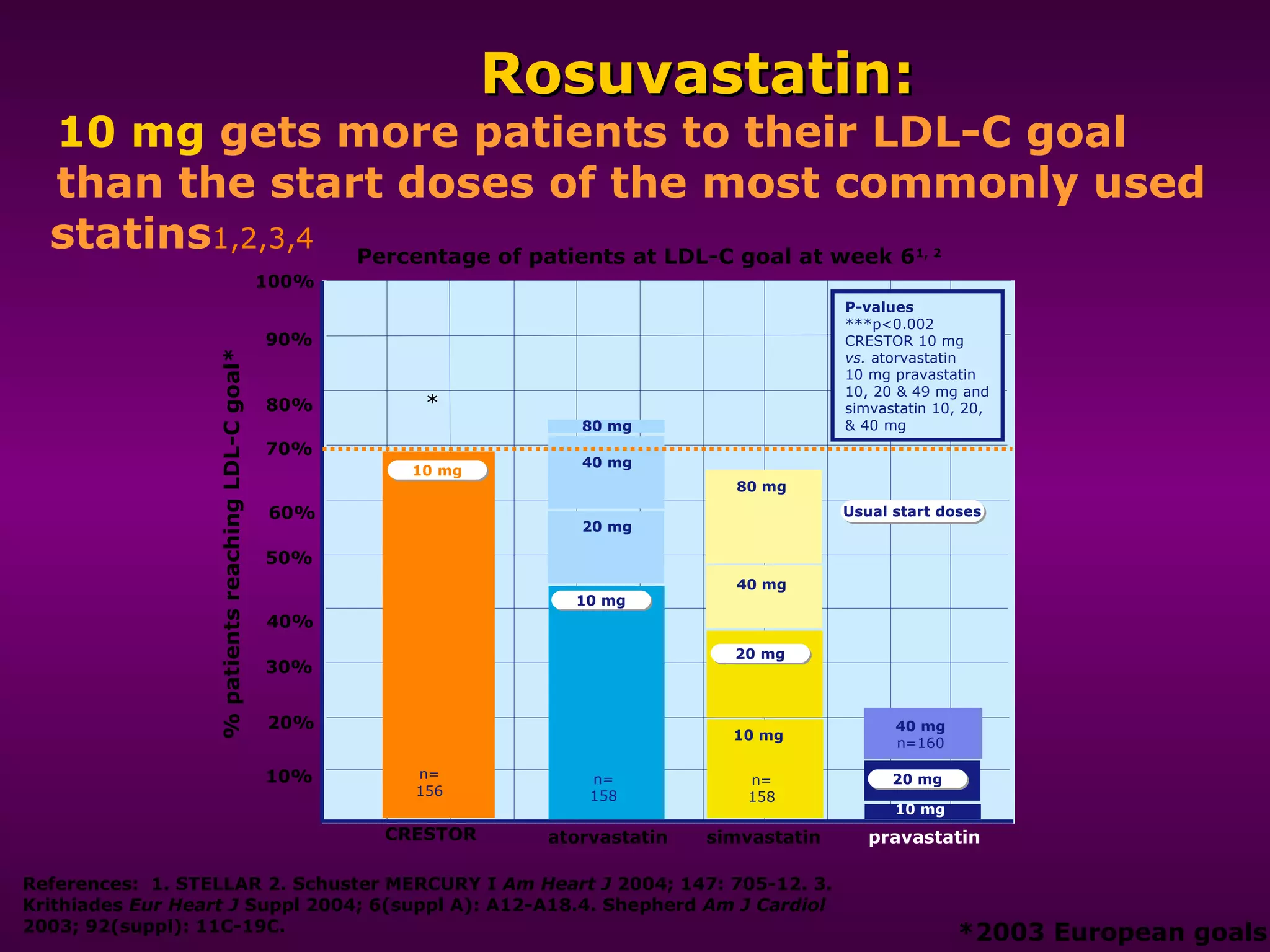
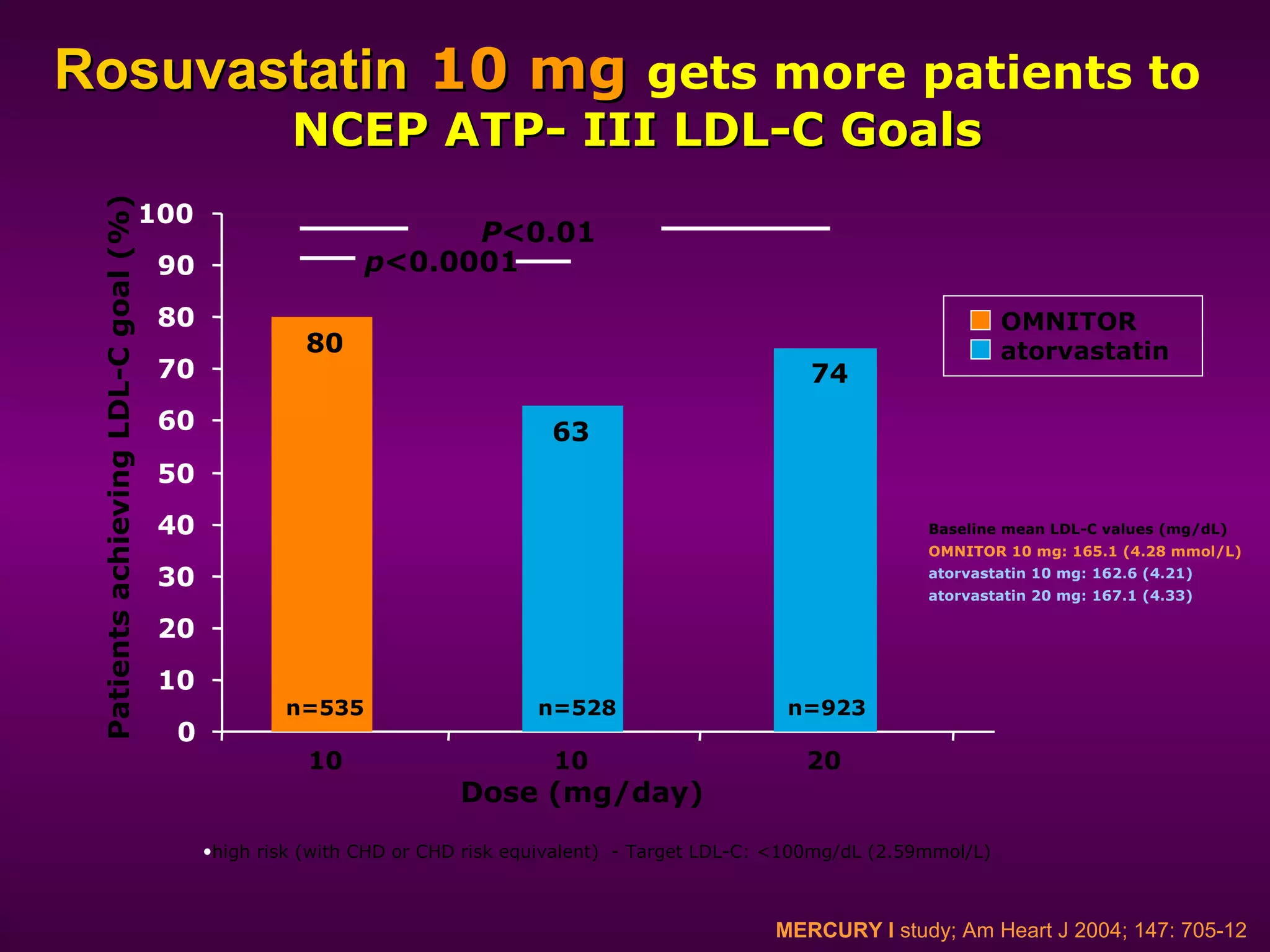



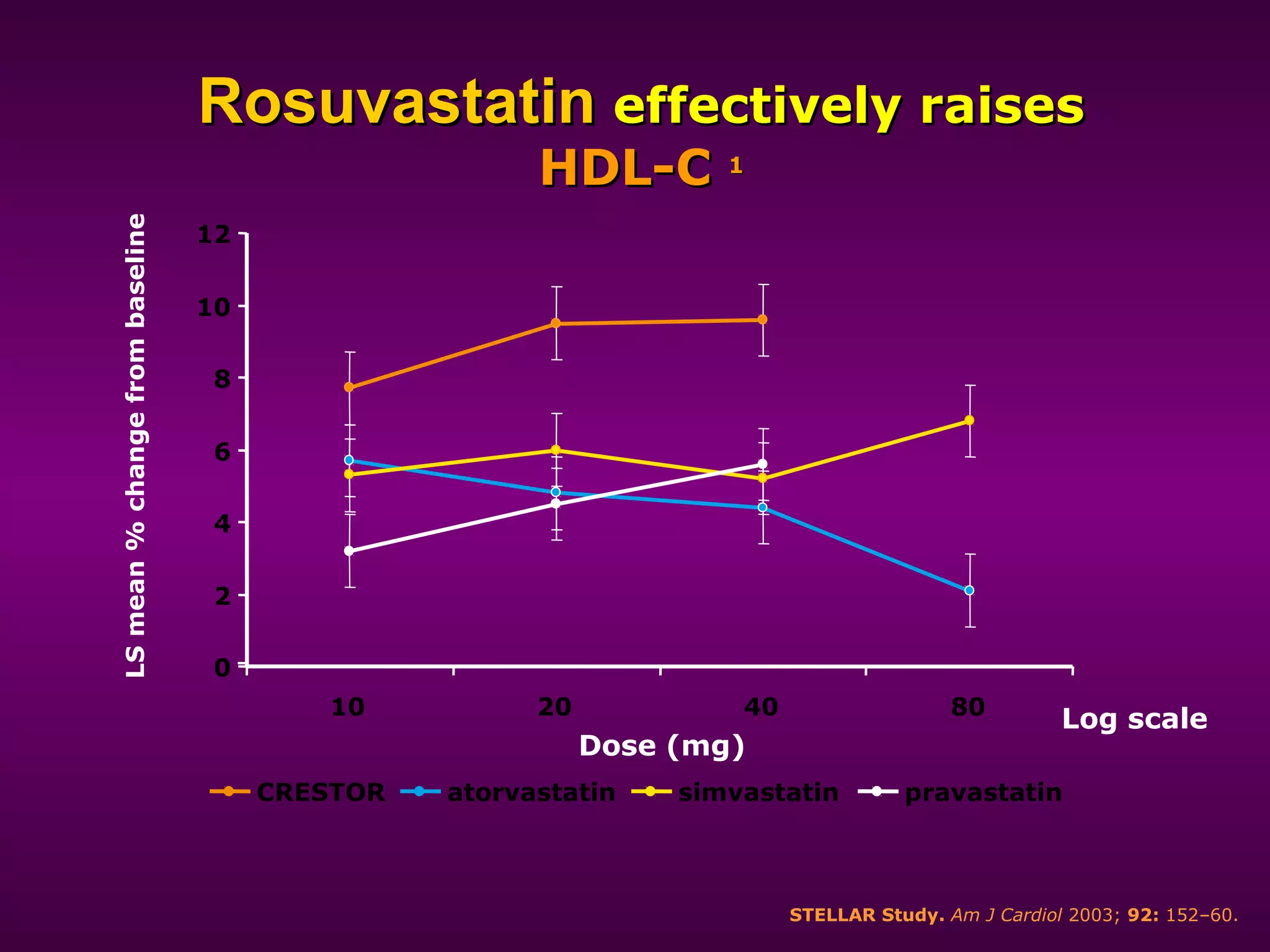

















The document discusses dyslipidemia and cardiovascular disease. It notes that cardiovascular disease is a leading cause of death worldwide and that dyslipidemia, specifically elevated LDL cholesterol, is a major modifiable risk factor. Statins are very effective at lowering LDL cholesterol levels and reducing cardiovascular events, with rosuvastatin being one of the statin drugs discussed as a new treatment option for dyslipidemia.























































































