Downloaded 1,967 times















































































































![Chapter 3.5 – Analgesics and Antipyretics
GM Hamad
product formed is para isobutyl phenyl propanoic acid / Ibuprofen.
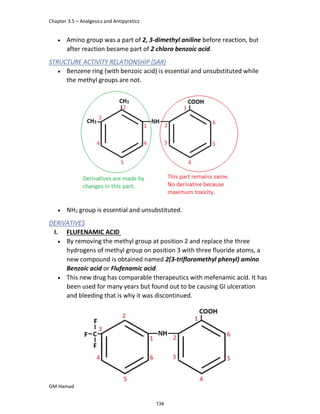
STRUCTURE ACTIVITY RELATIONSHIP (SAR)
In ibuprofen 2 groups are attached at position 1 and 4.
At position 1, there is alkyl group with any other group, therapeutic
activity will be terminated.
At position 4, there is carboxylic acid functional group which is propionic
acid, in combinational chemistry, propionic acid was substituted by
acetic acid which leads to the formation of Ibufenac which can be used
therapeutically but have increased therapeutic effects.
ISOMERS
It is both isomers are therapeutically and optically active.
But the racemic mixture is therapeutically active but optically inactive.
EUDISMIC RATIO
It is defined as a comparison of 2 enantiomers of drugs in
pharmacological activity.
Ibuprofen is usually marketed as a racemic mixture (50:50 mixture of
[S],[+] and [R],[-] enantiomers), although the [S],[+] enantiomer is more
active and hence has increased anti-inflammatory activity.
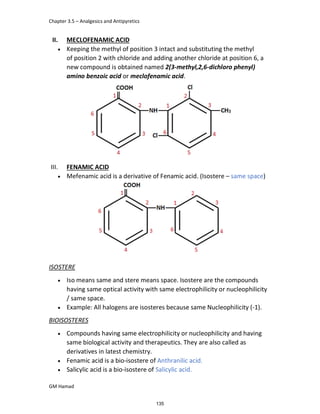
DERIVATIVE
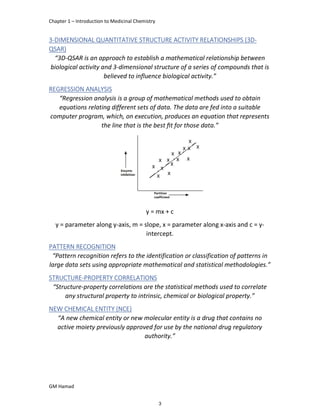
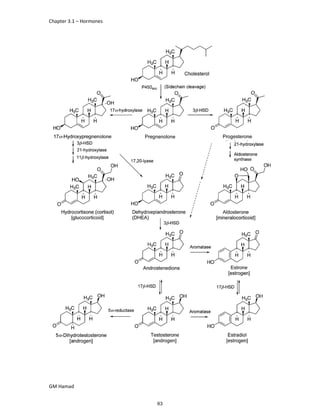
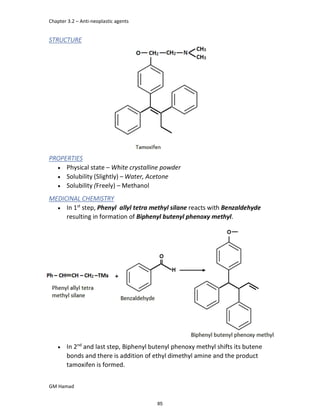
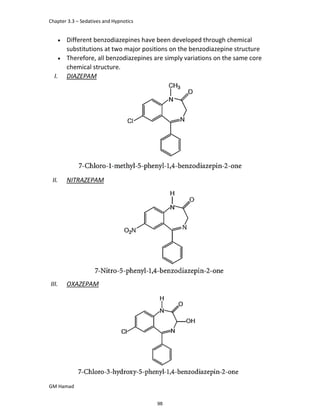
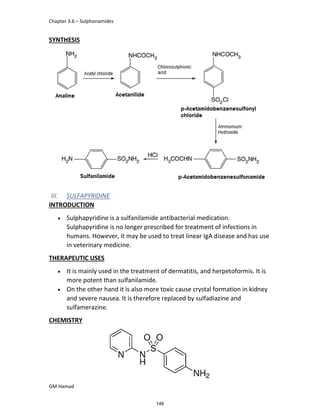
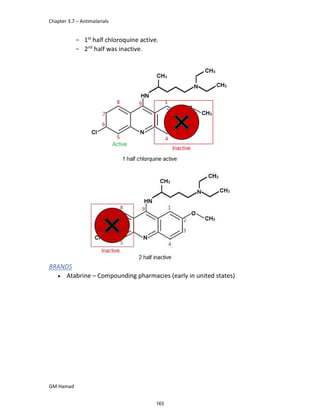
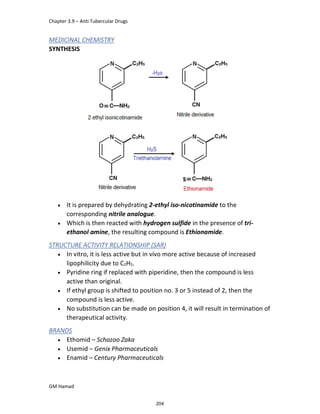
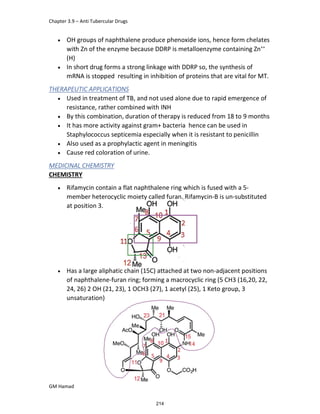
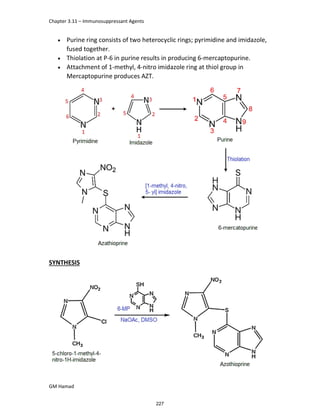
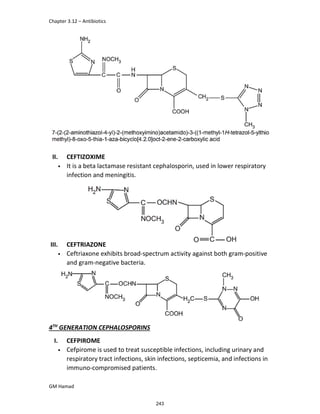
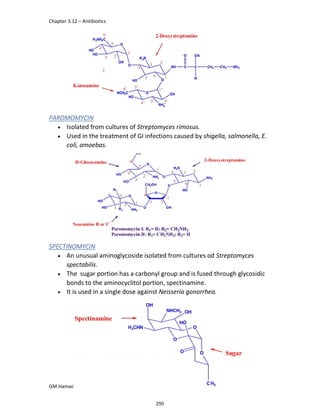
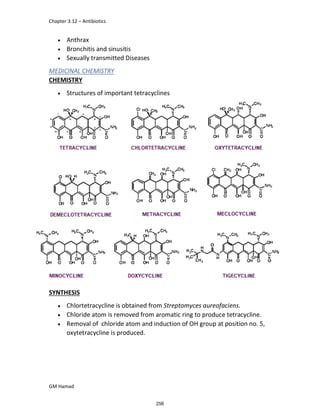
IBUFENAC
Ibufenac is mono carboxylic acid
that is acetic acid.
It was effective in treatment of
Rheumatoid arthritis but was
discontinued due to hepatotoxic
effects.
128](https://image.slidesharecdn.com/gmnotesmedicinalchemistry-221109090226-ccdf1c86/85/Medicinal-Chemistry-Complete-Notes-130-320.jpg)








![Chapter 3.5 – Analgesics and Antipyretics
GM Hamad
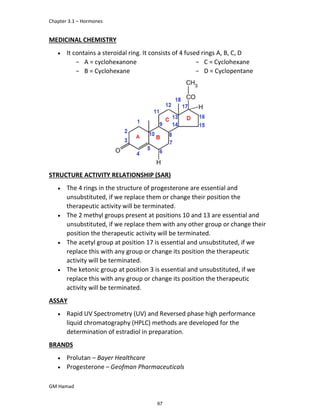
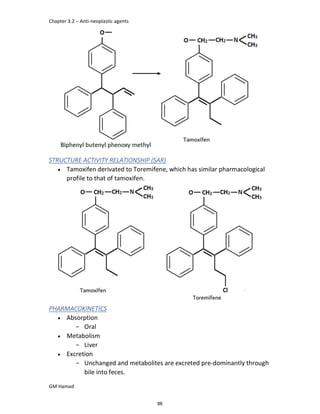
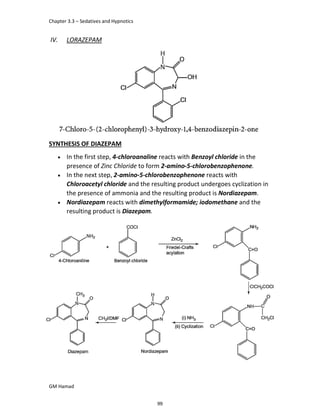
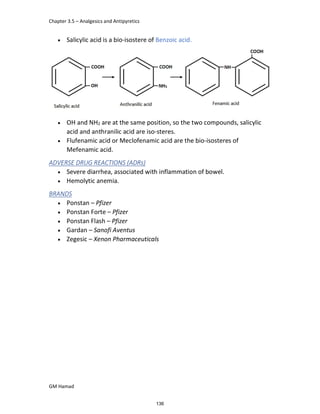
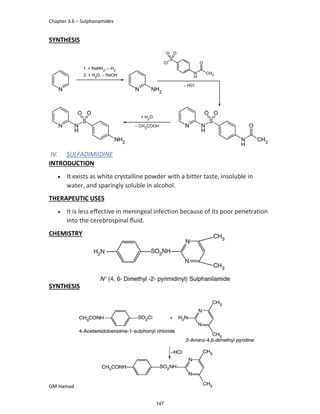
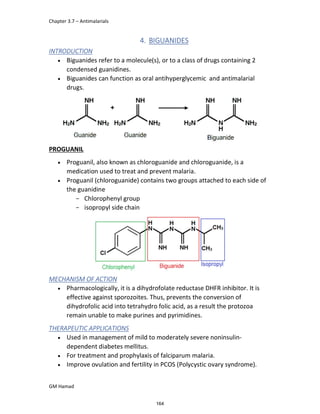
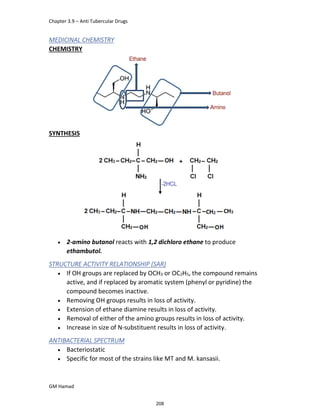
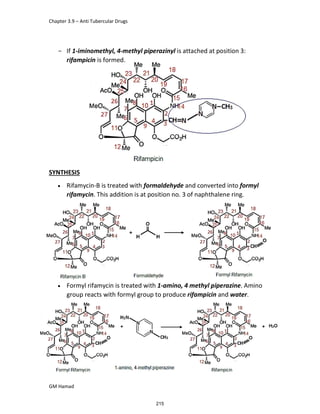
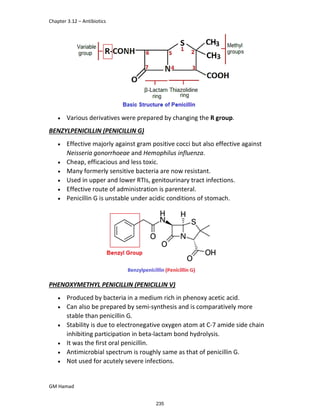
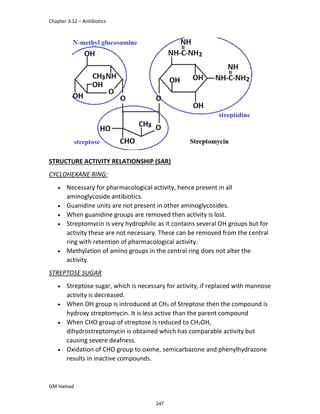
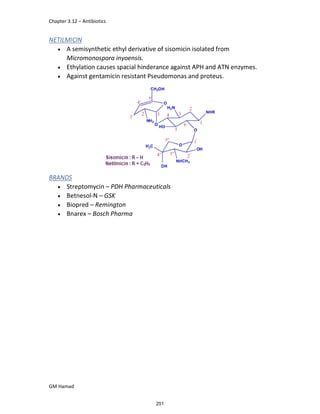
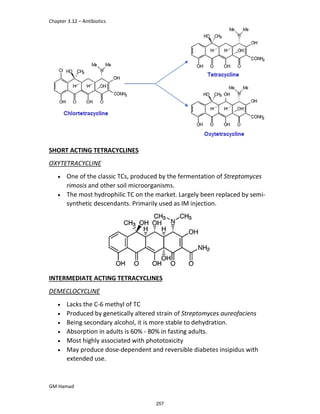
In the next step, there is electrophilic substitution reaction at position
no. 2, the product formed is 2 methoxy 6 bromo naphthalene / 6 bromo
2 methoxy naphthalene.
In the next step, there is substitution of bromine by propionic acid, the
product formed is 2 methoxy naphthalene 6 propionic acid / Naproxen
that is attached to carbon 2.
]
In the last step, naproxen is reacted with NaOH to form atypical salt i.e.,
2 methoxy naphthalene 6 sodium propionate / Naproxen sodium.
Salt is formed to increase bioavailability and solubility.
142](https://image.slidesharecdn.com/gmnotesmedicinalchemistry-221109090226-ccdf1c86/85/Medicinal-Chemistry-Complete-Notes-144-320.jpg)









































































![Medicinal Viva Questions
GM Hamad Bisma Mushtaq
11.Define Azine and Azole.
PRACTICALS
1. Definition of molarity, molality, normality, gram equivalent.
2. What is ester?
3. What is amine and amide?
4. What is standard solution?
5. What is thermolysis?
6. How to make 1N solution of Hcl and H2SO4?
7. What is hydrogenation, dehydration, oxidation, reduction?
8. What is valency of N, O, S?
9. What is meant by pseudo?
10.Where are mast cells present?
11.What is nascent hydrogen [H]?
12.Definition of titration, types, reason?
13.What is titer, analyte, titrates?
14.What are the derivatives of acetaminophen? Why only paracetamol is
used?
15.Give mechanism of disprin in blood thinning?
16.What is cyanide? Why is it toxic?
17.Give uses of paracetamol?
18.Structure of morphine, codeine, pholcodine, salol?
19.What are chemical names of ester and ether?
20.How is oil of winter green isolated from plants?
21.Physical state of methyl salicylate?
22.Brand names of different products containing methyl salicylate.
23.Function of H2SO4 in a chemical reaction?
24.Uses of 5-amino salicylic acid? (IBS)
25.Amine, amide, ester drugs?
268](https://image.slidesharecdn.com/gmnotesmedicinalchemistry-221109090226-ccdf1c86/85/Medicinal-Chemistry-Complete-Notes-270-320.jpg)


This document provides an overview of key concepts in medicinal chemistry. It defines important terms like drug, drug design, drug discovery, drug development, lead compounds, structure activity relationships, quantitative structure activity relationships, molecular targets, and drug-likeness. It also describes concepts such as privileged structures, pharmacophores, analogues, attrition rates, back-up compounds, best-in-class drugs, bioassays, bioinformatics, chemical databases, and chemical libraries. The document serves as an introduction to medicinal chemistry for students in a Doctor of Pharmacy program.




















![ANTIBIOTICS[PENICILLINS] MEDICINAL CHEMISTRY BY RAVISANKAR](https://cdn.slidesharecdn.com/ss_thumbnails/penicillin-1-ppt-new-130616074411-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
























































