Downloaded 1,079 times











































































































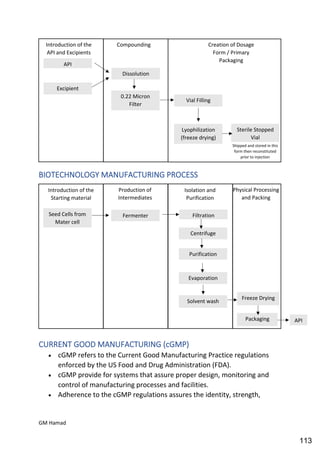










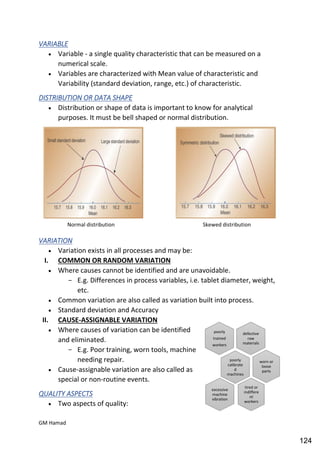
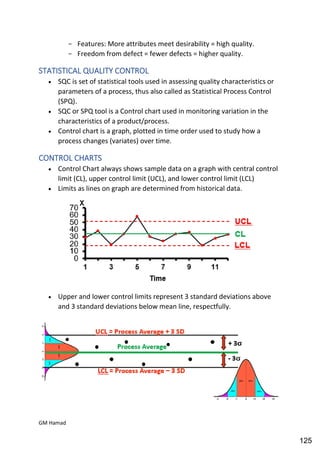

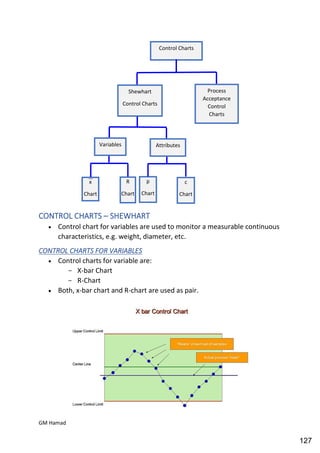







This document provides an overview of quality control procedures for pharmaceutical products. It discusses quality management systems, quality assurance, good manufacturing practices, good laboratory practices, and validation procedures. It also describes specific quality control tests for solid dosage forms like tablets, including tests for hardness, thickness, friability, weight variation, disintegration, and dissolution. The document provides details on the apparatus and procedures used for each quality control test of tablets.






































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)







