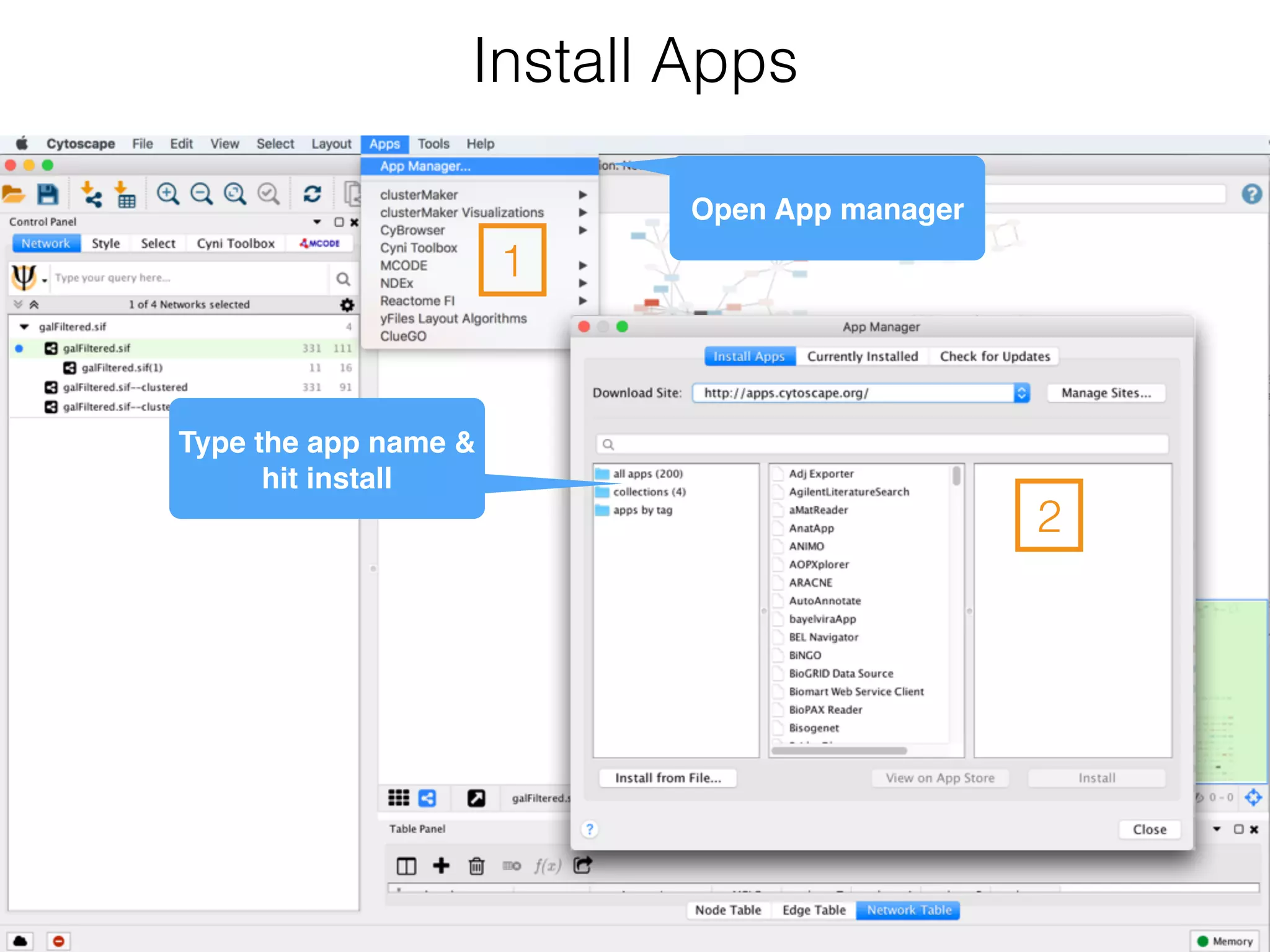
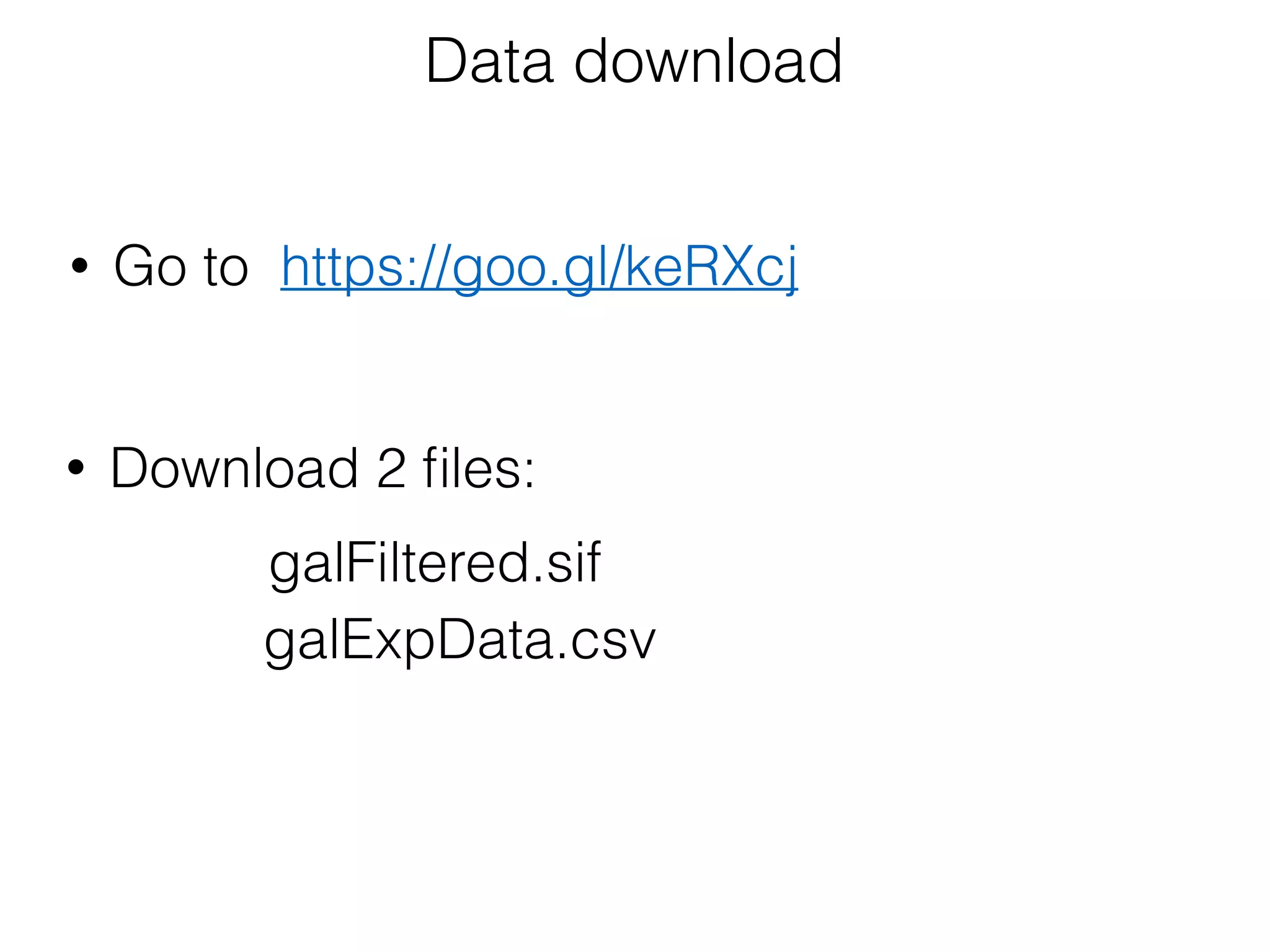
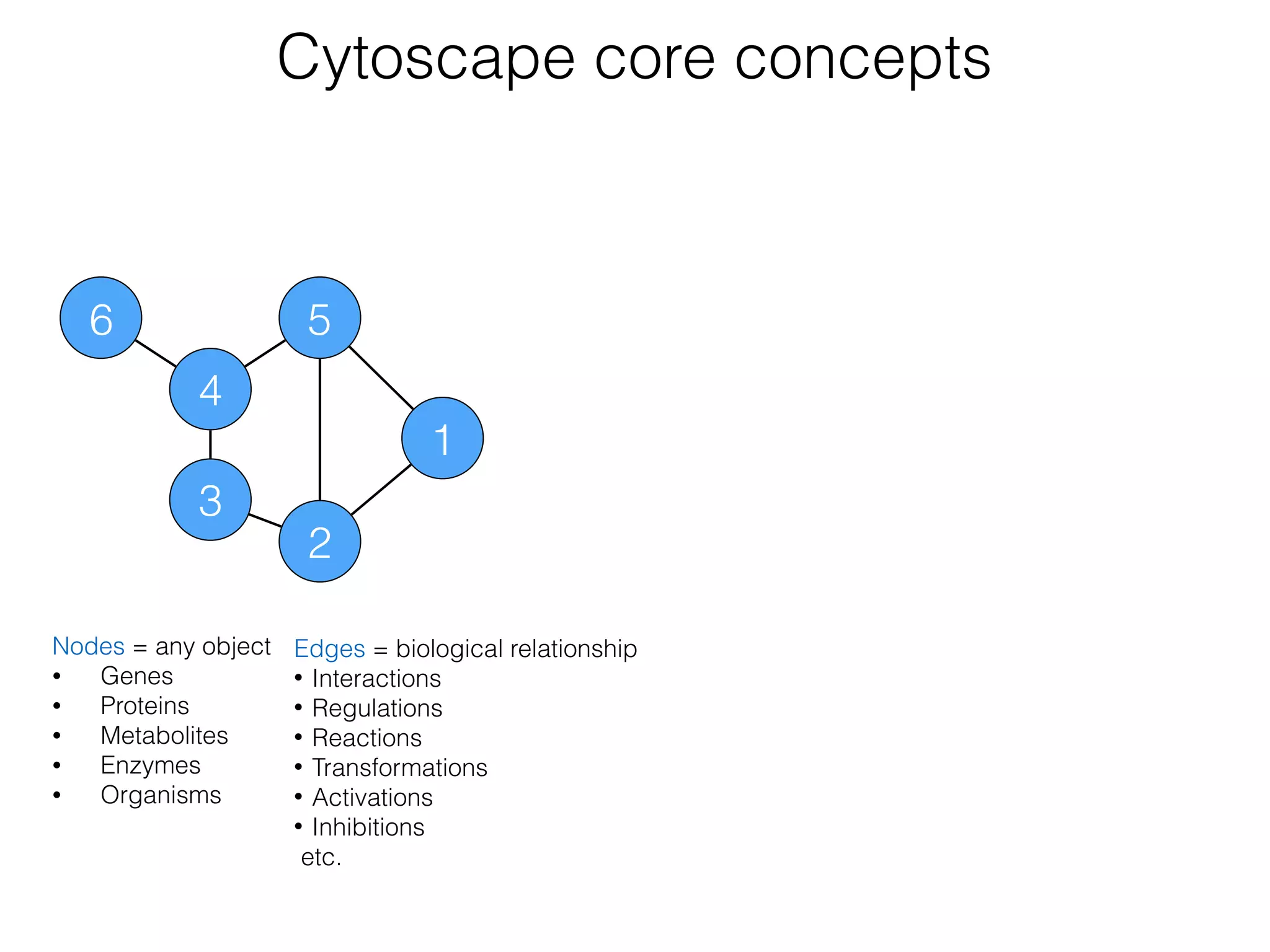
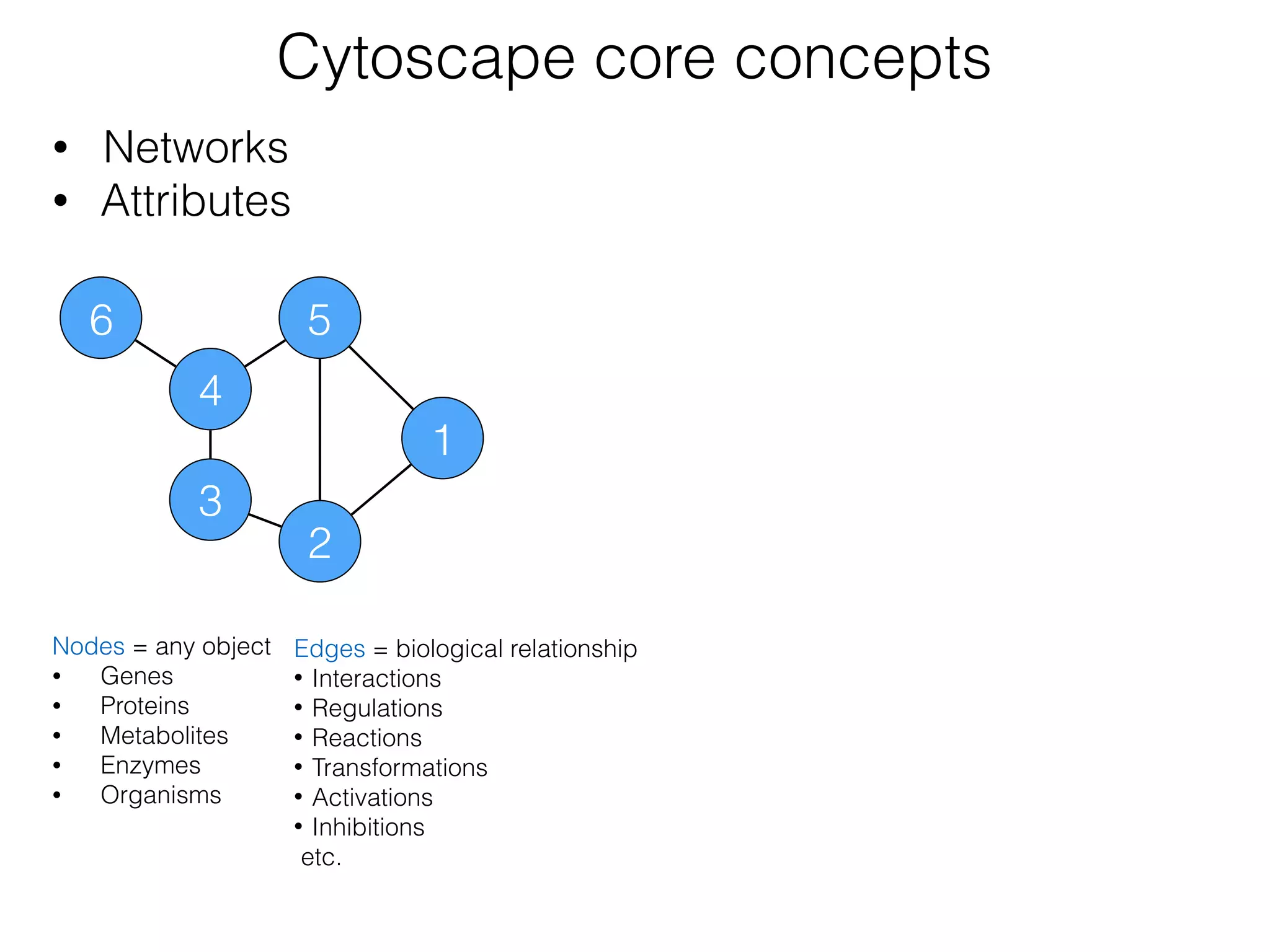
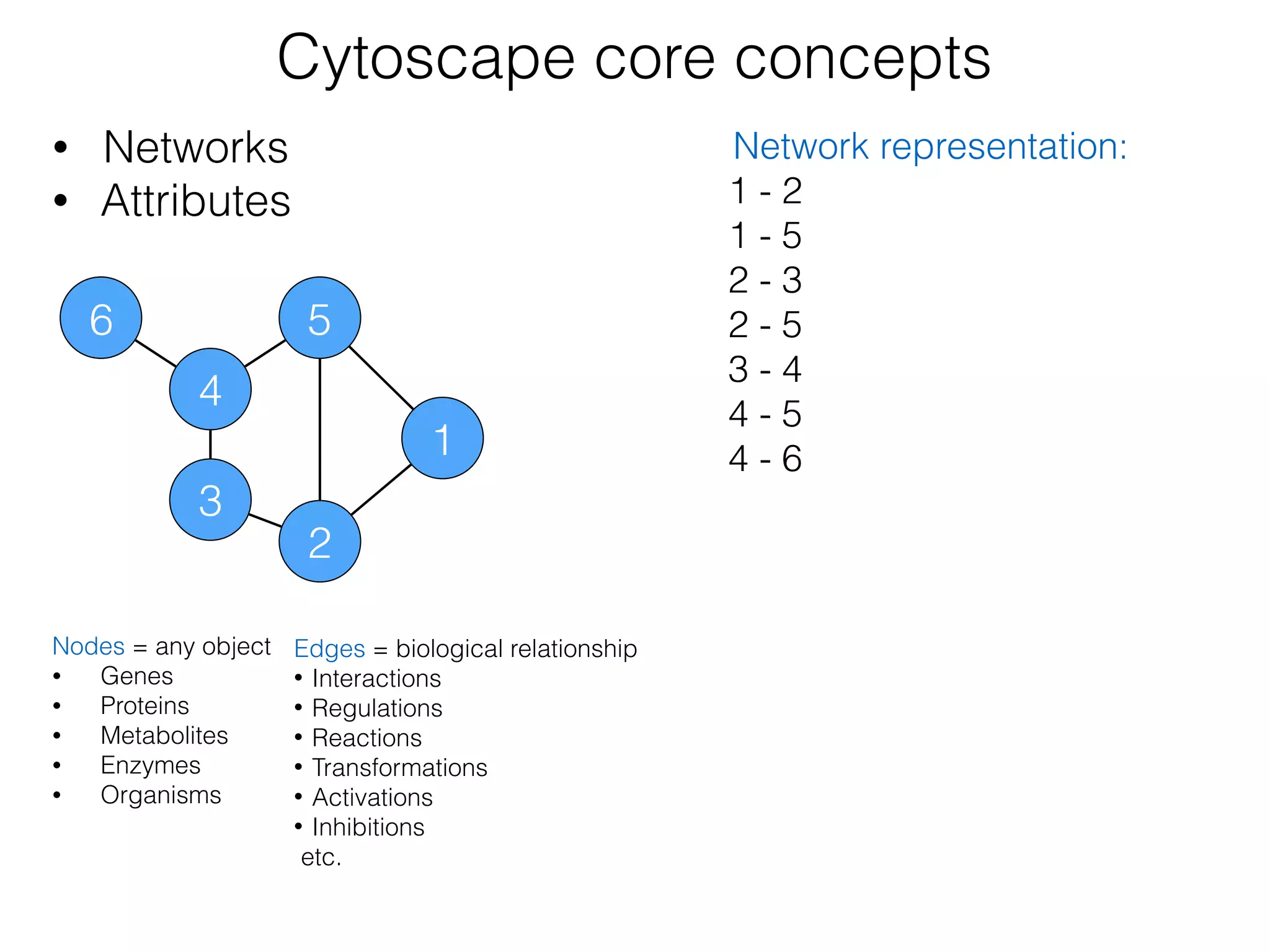
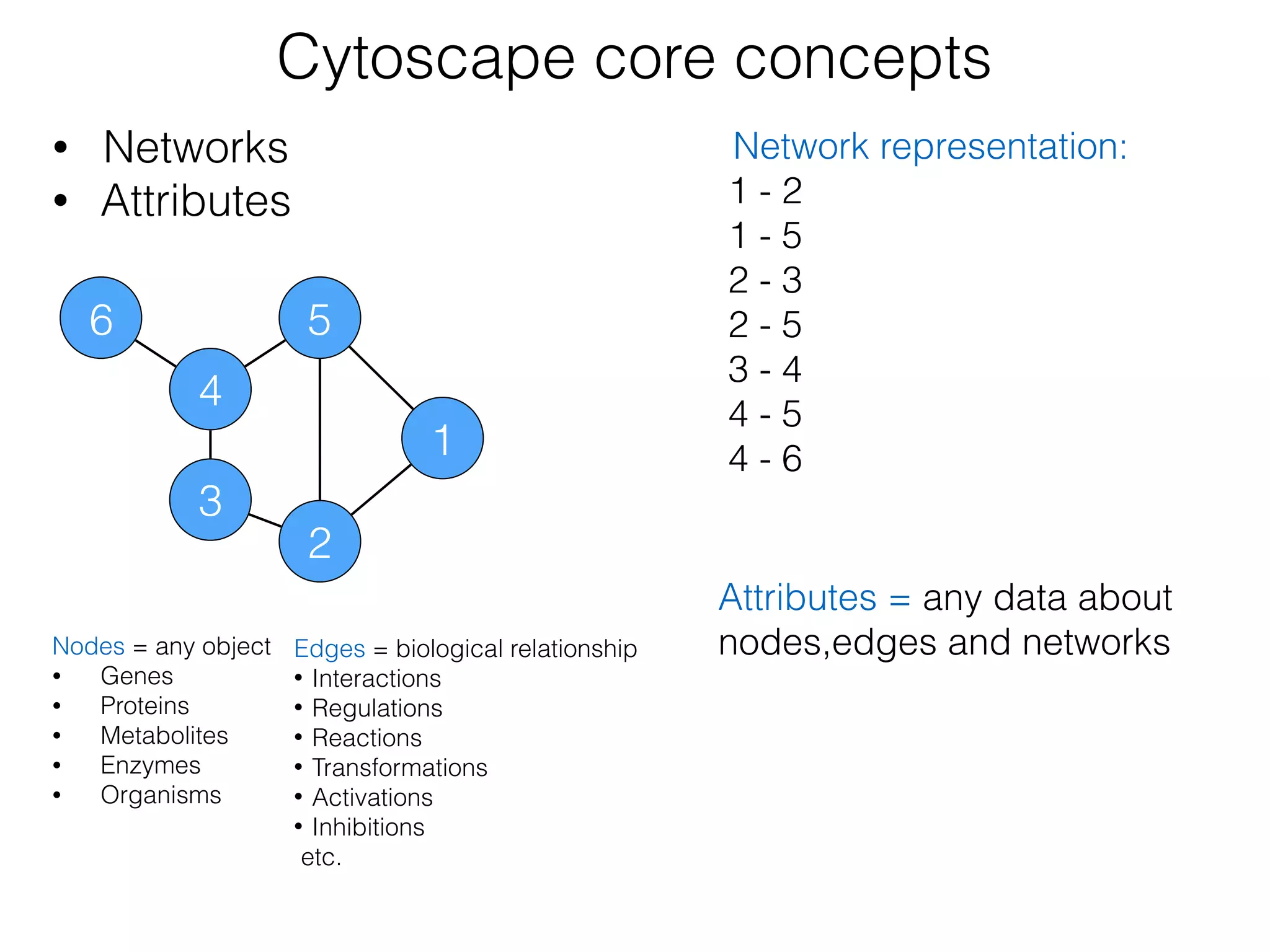
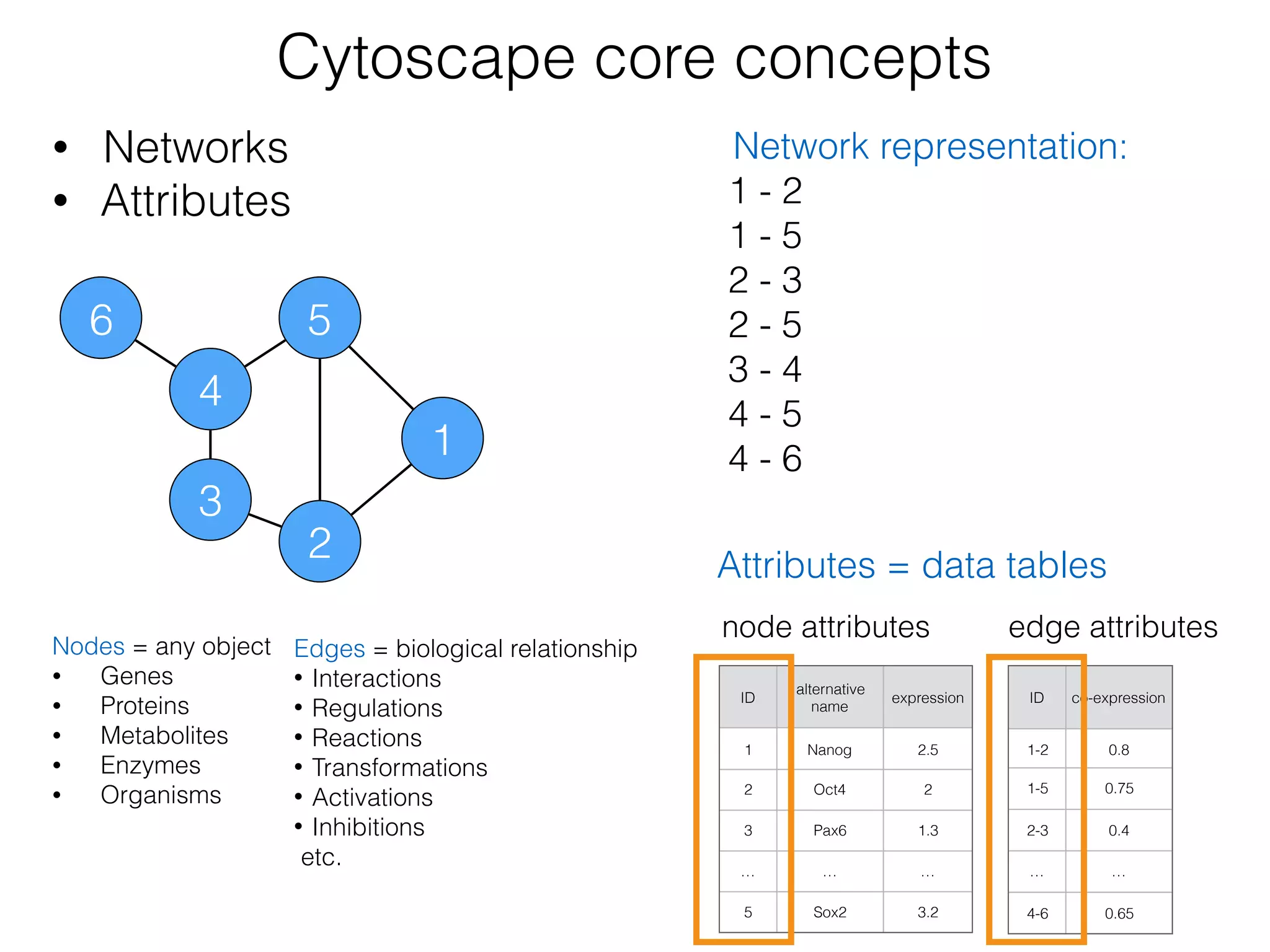
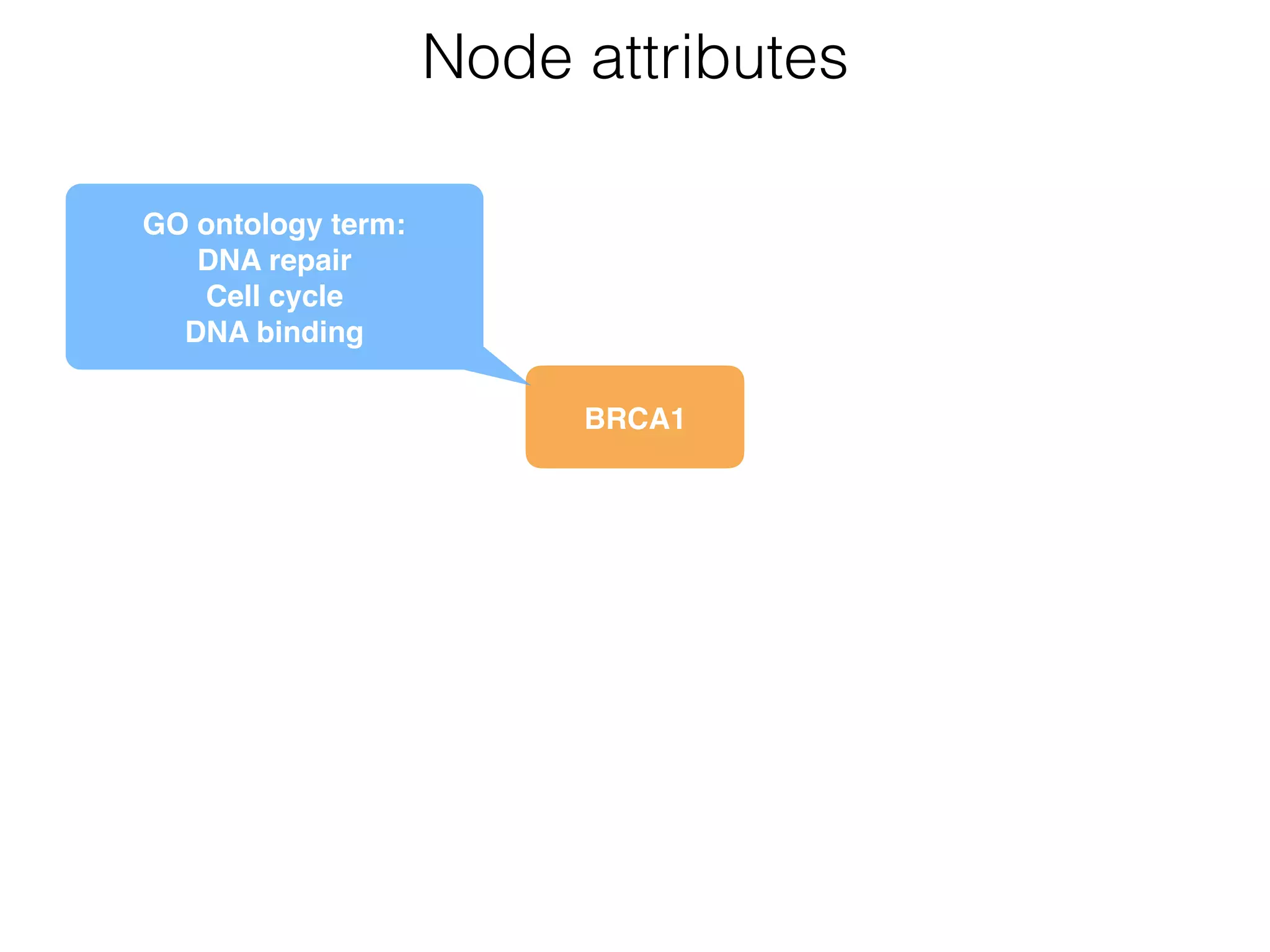
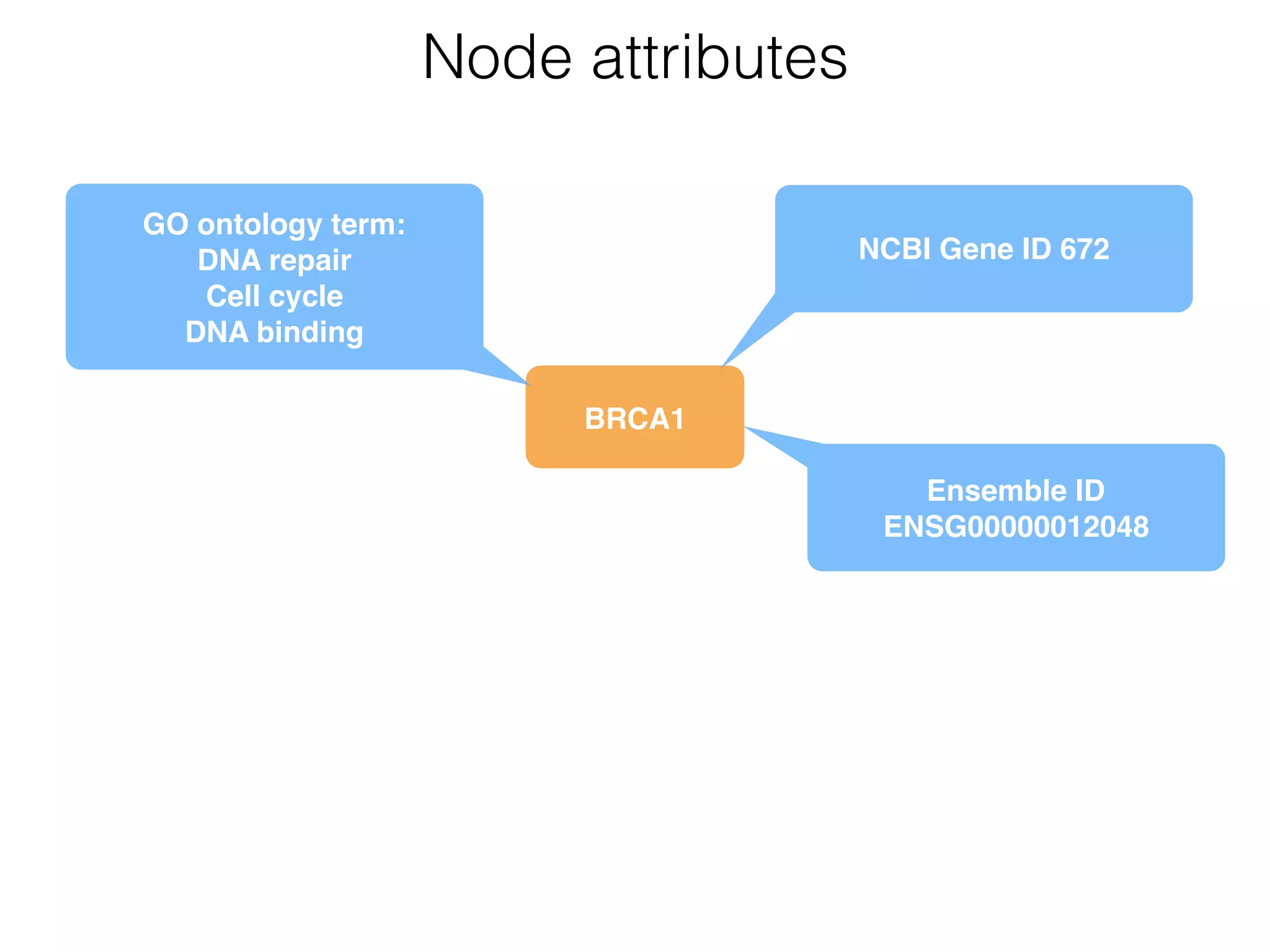
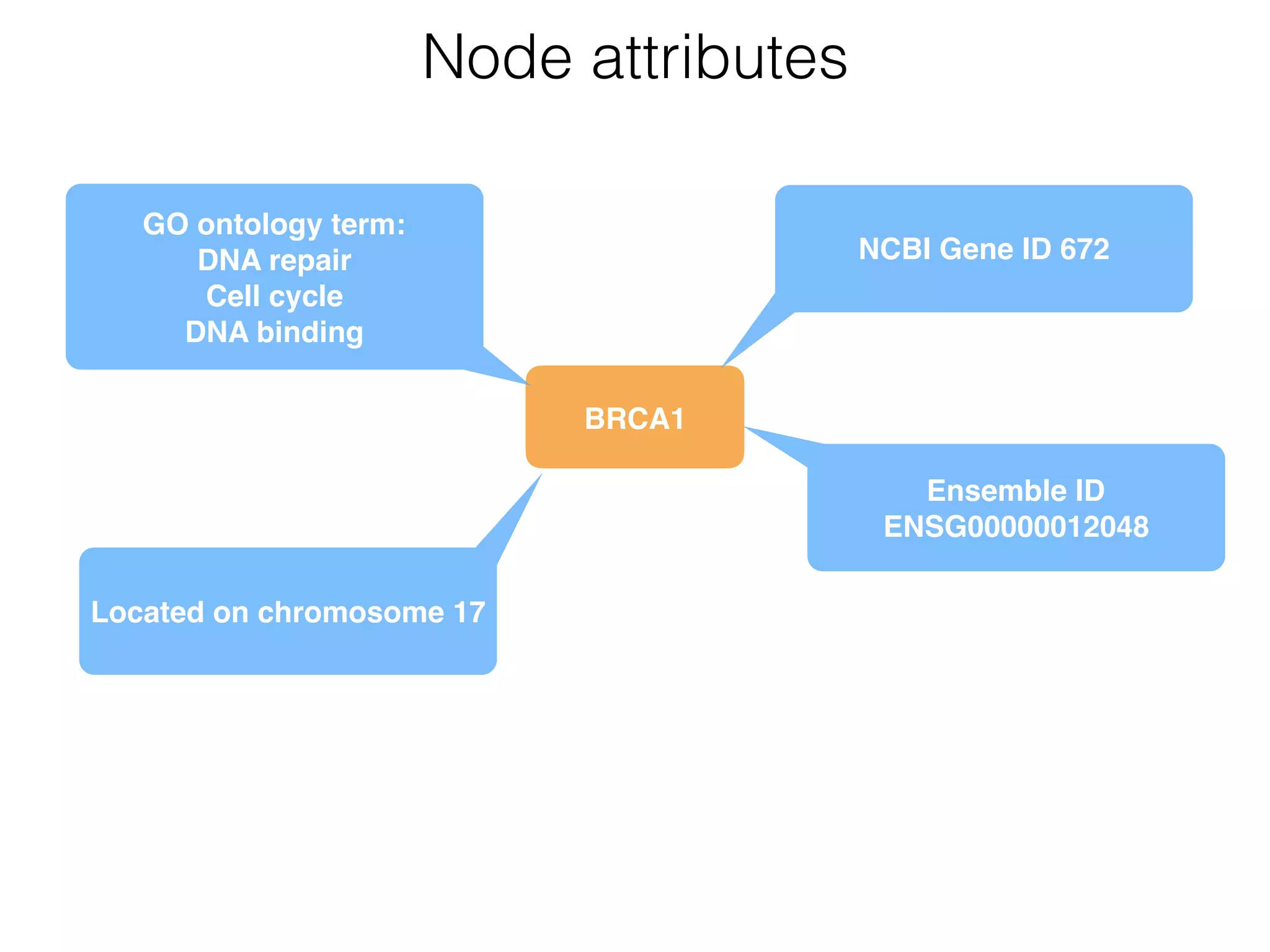
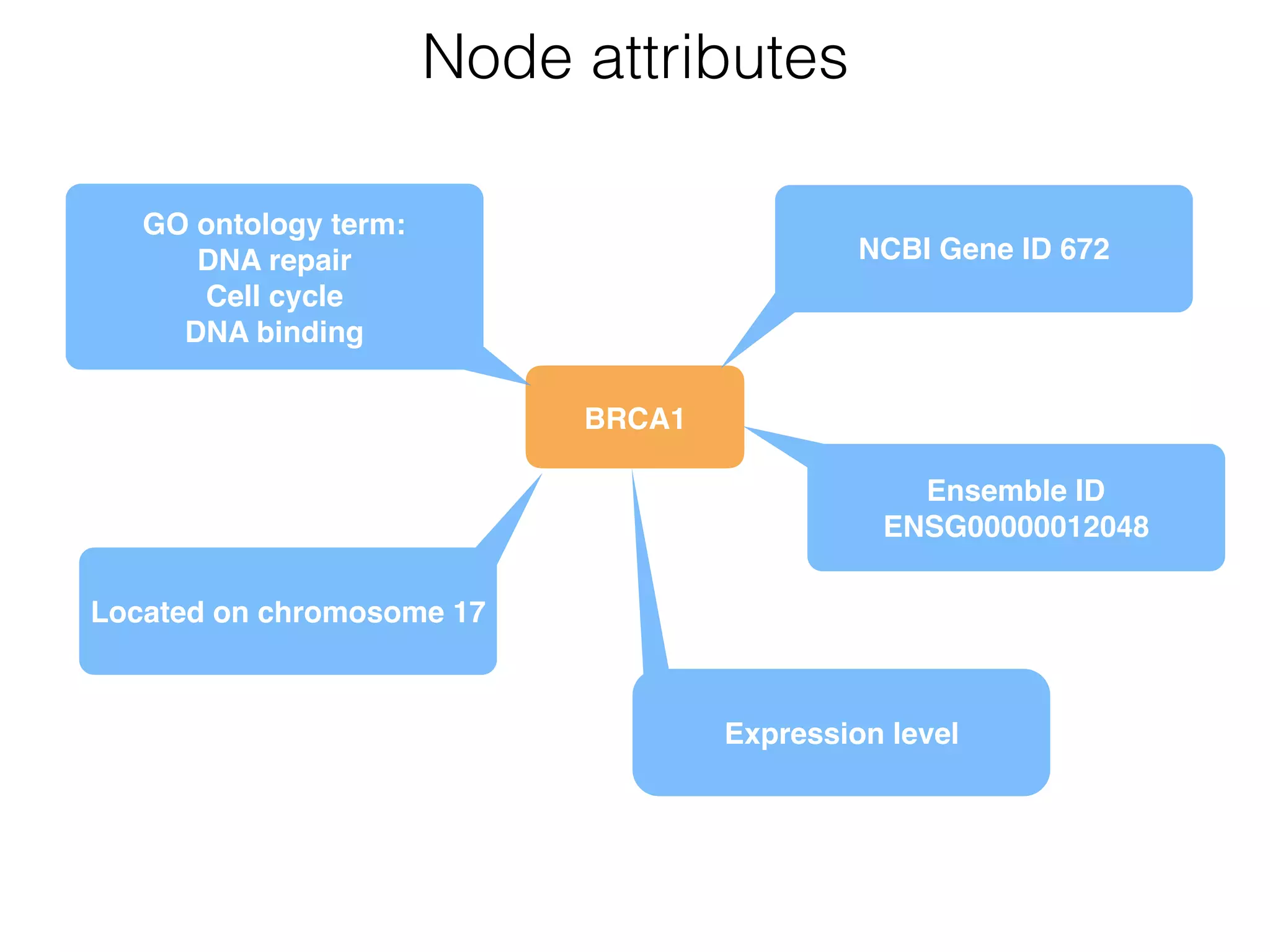
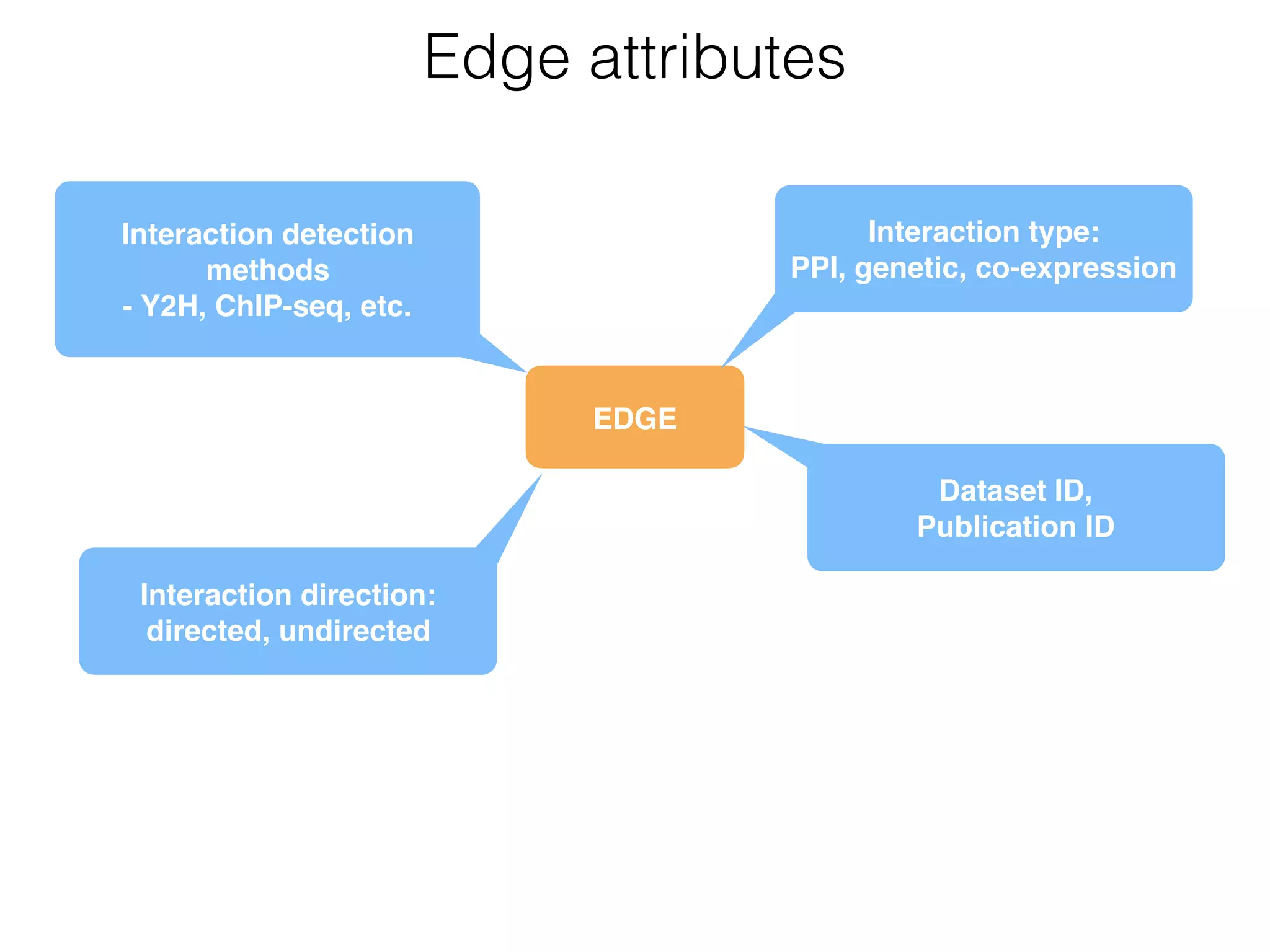
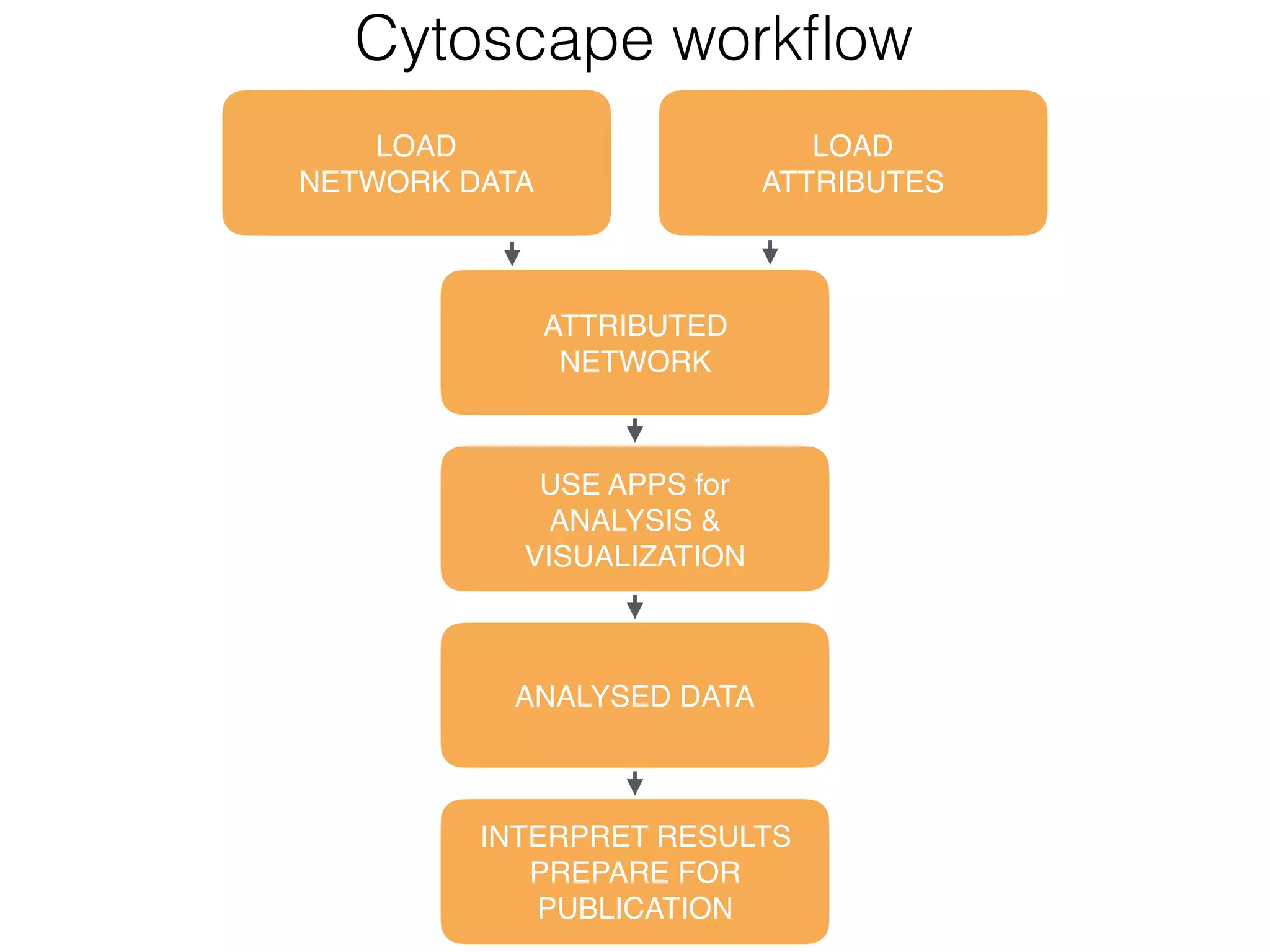
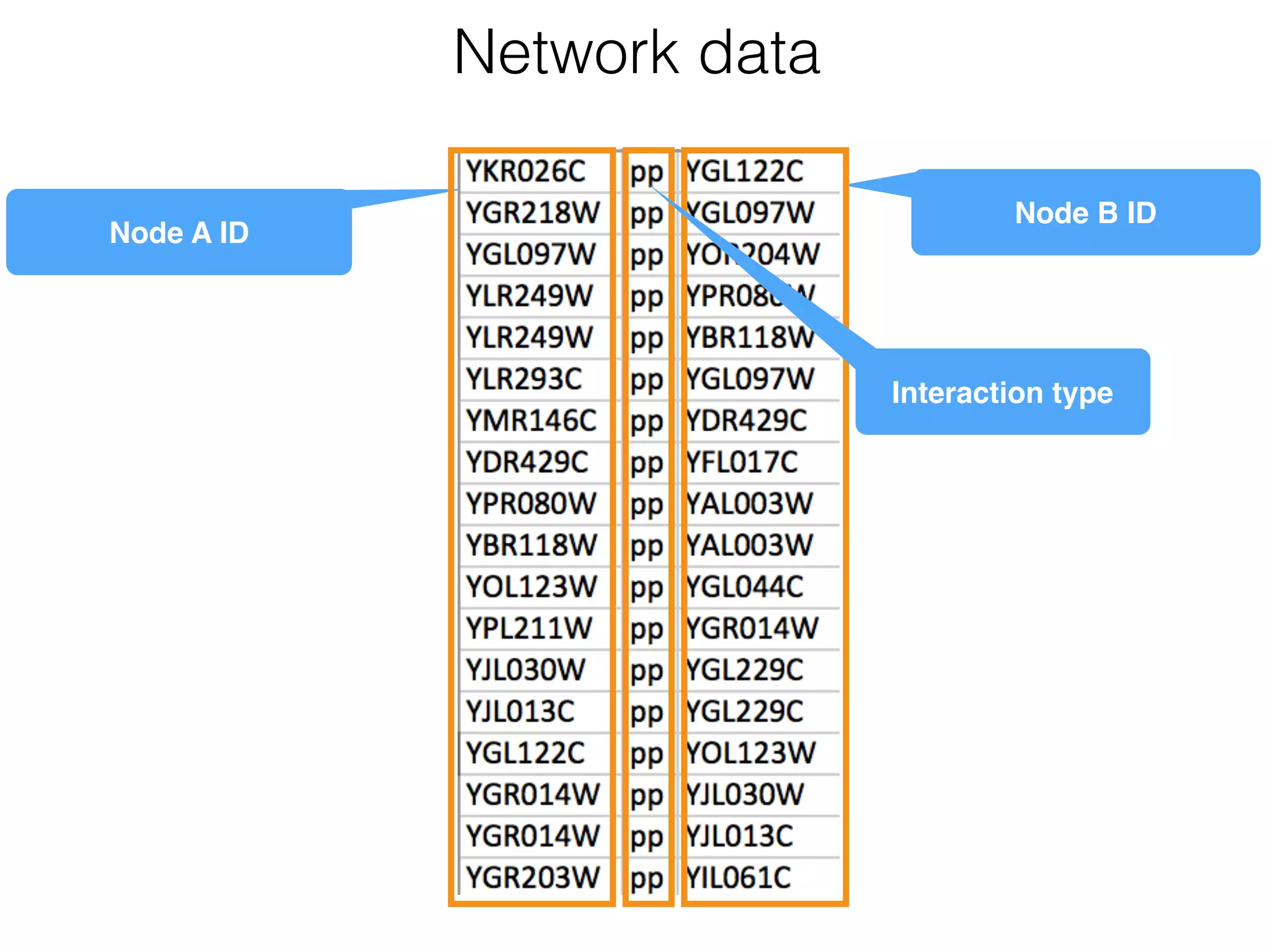
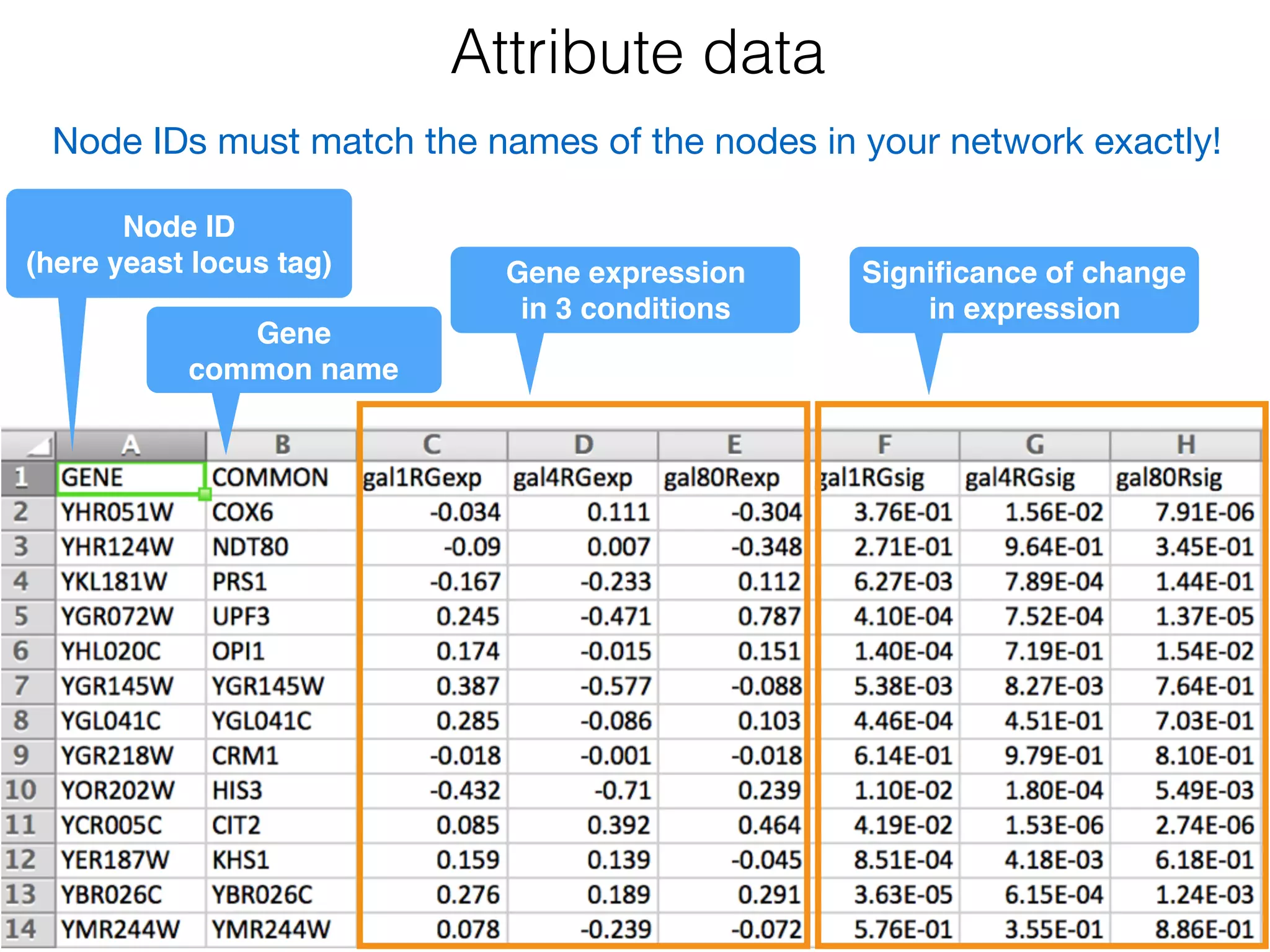
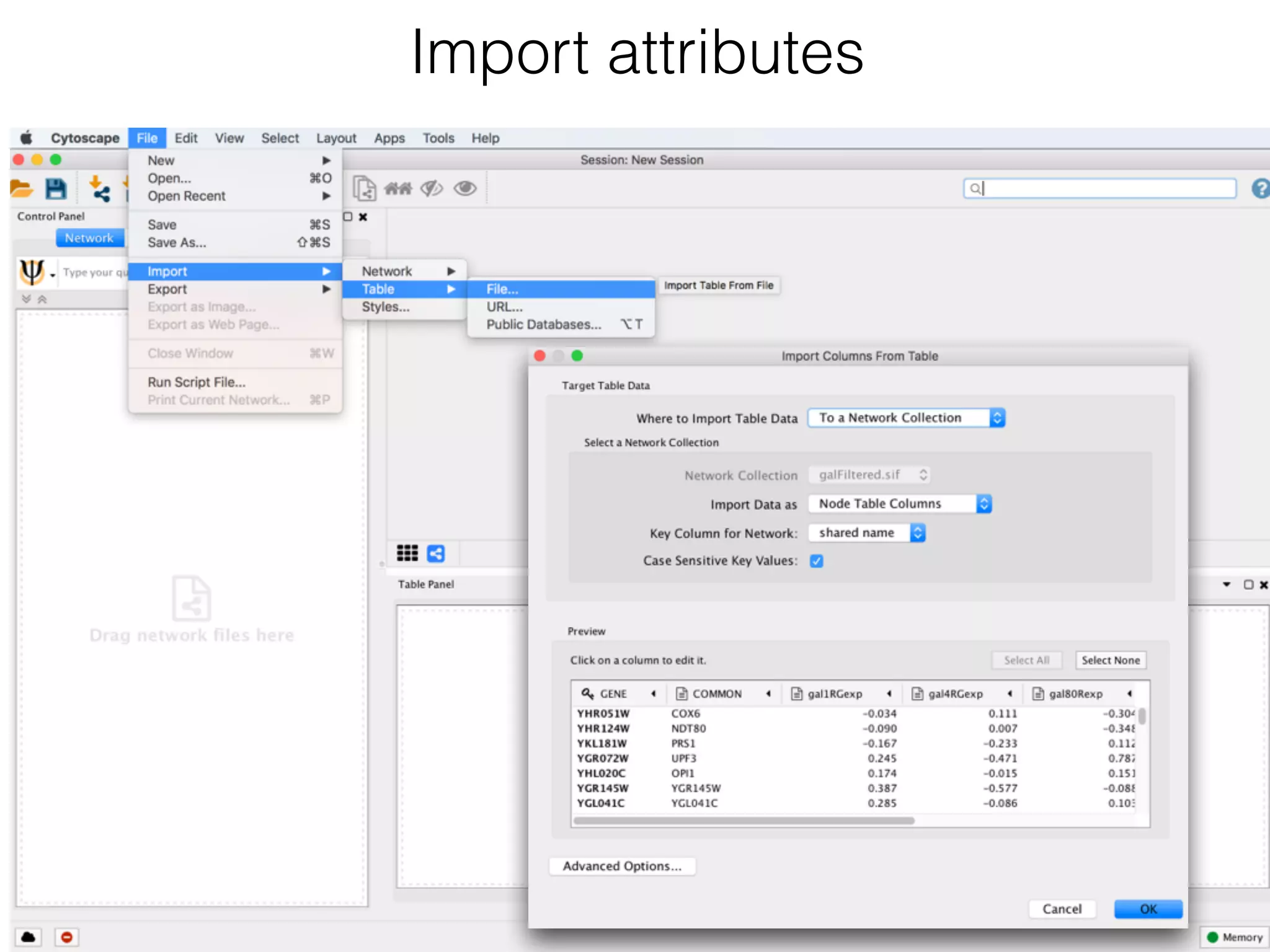
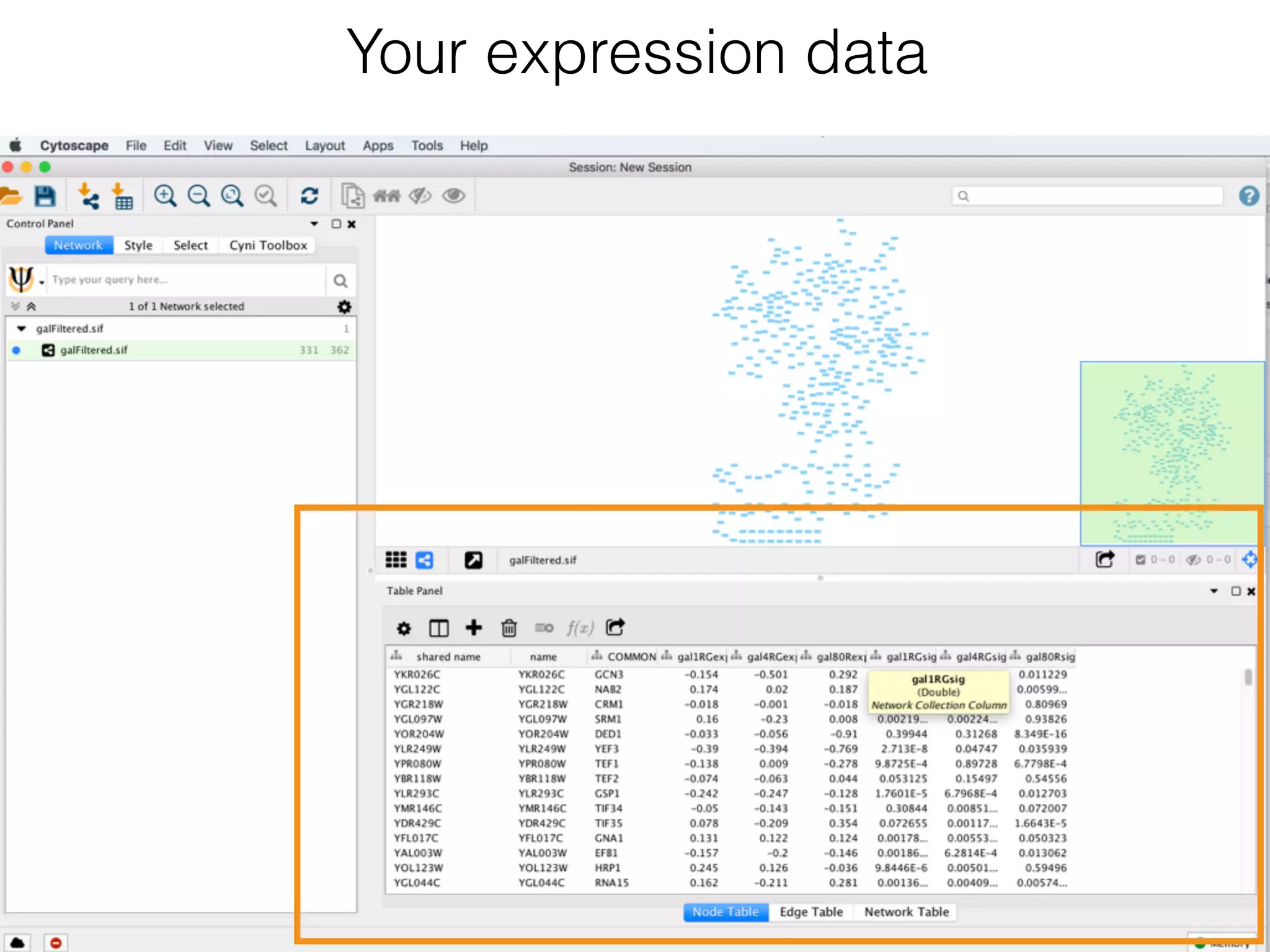
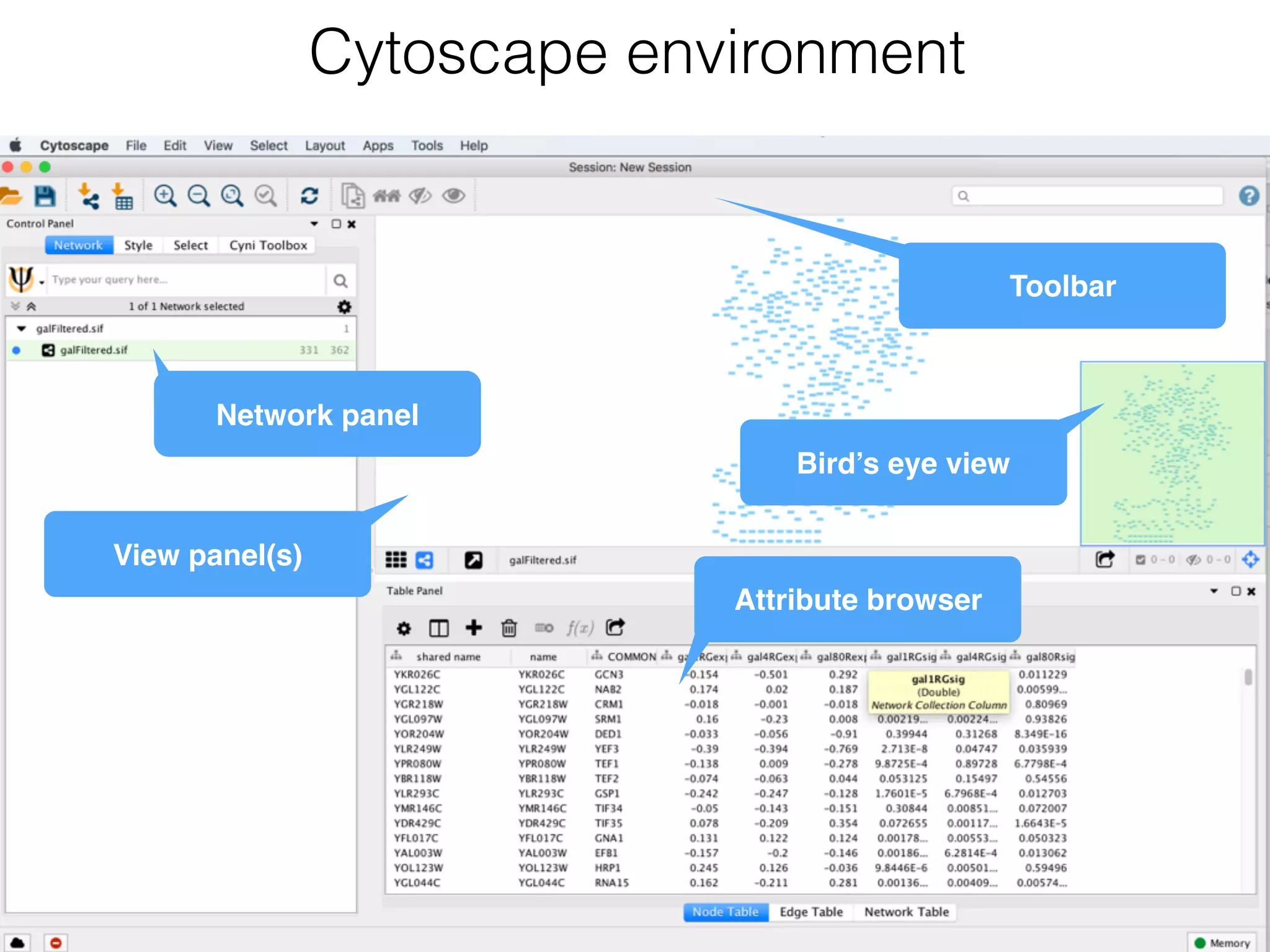
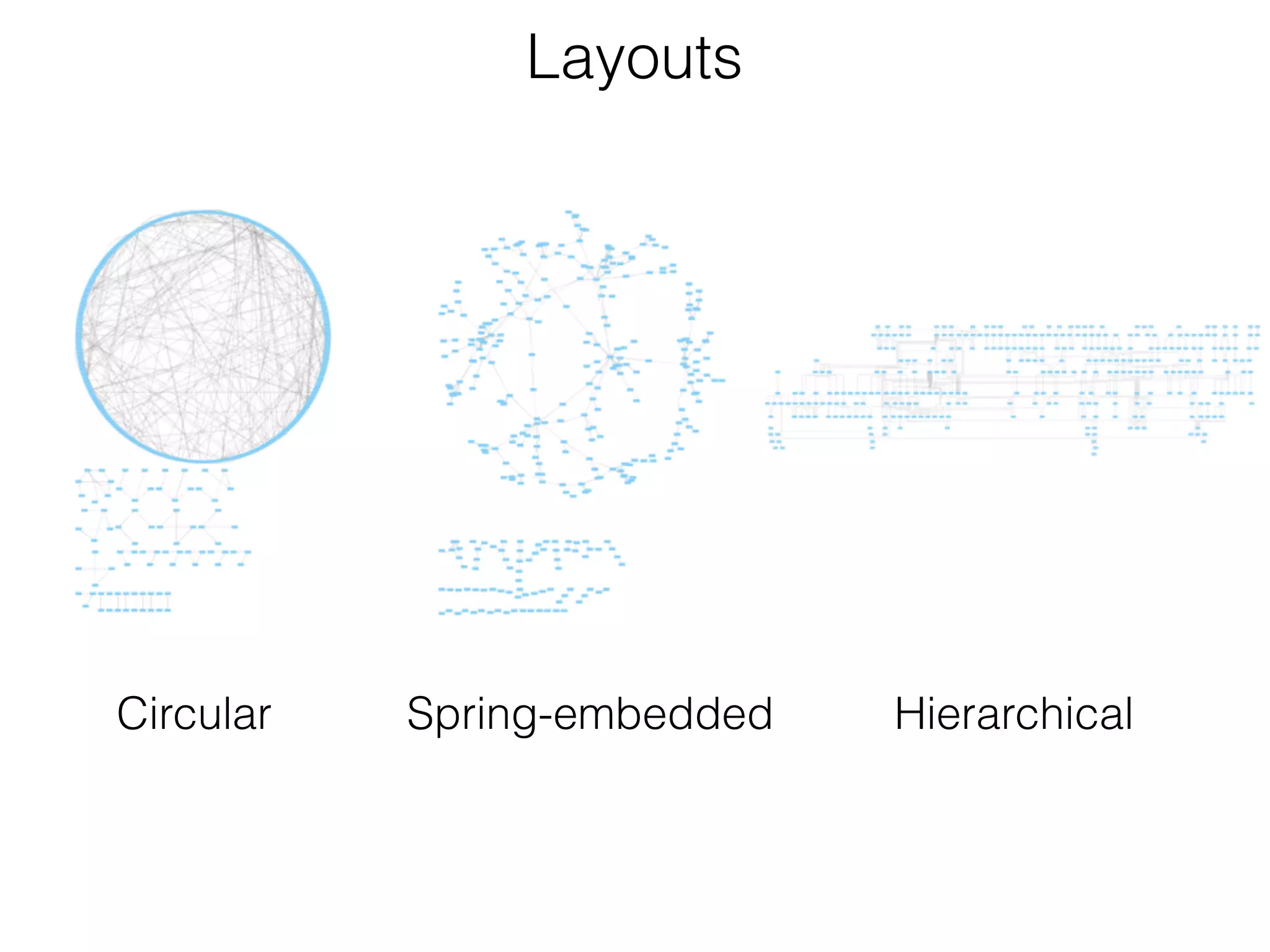
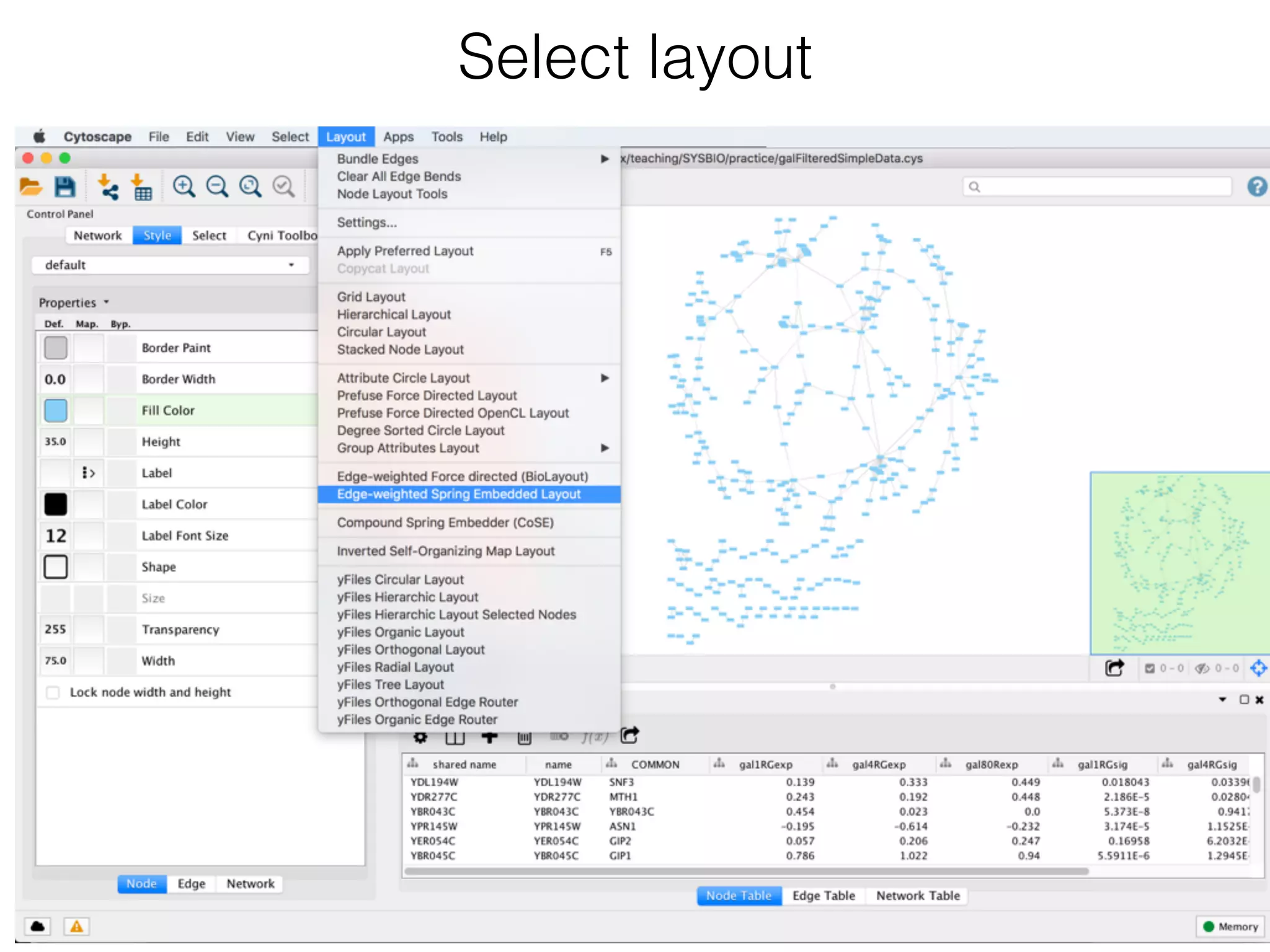
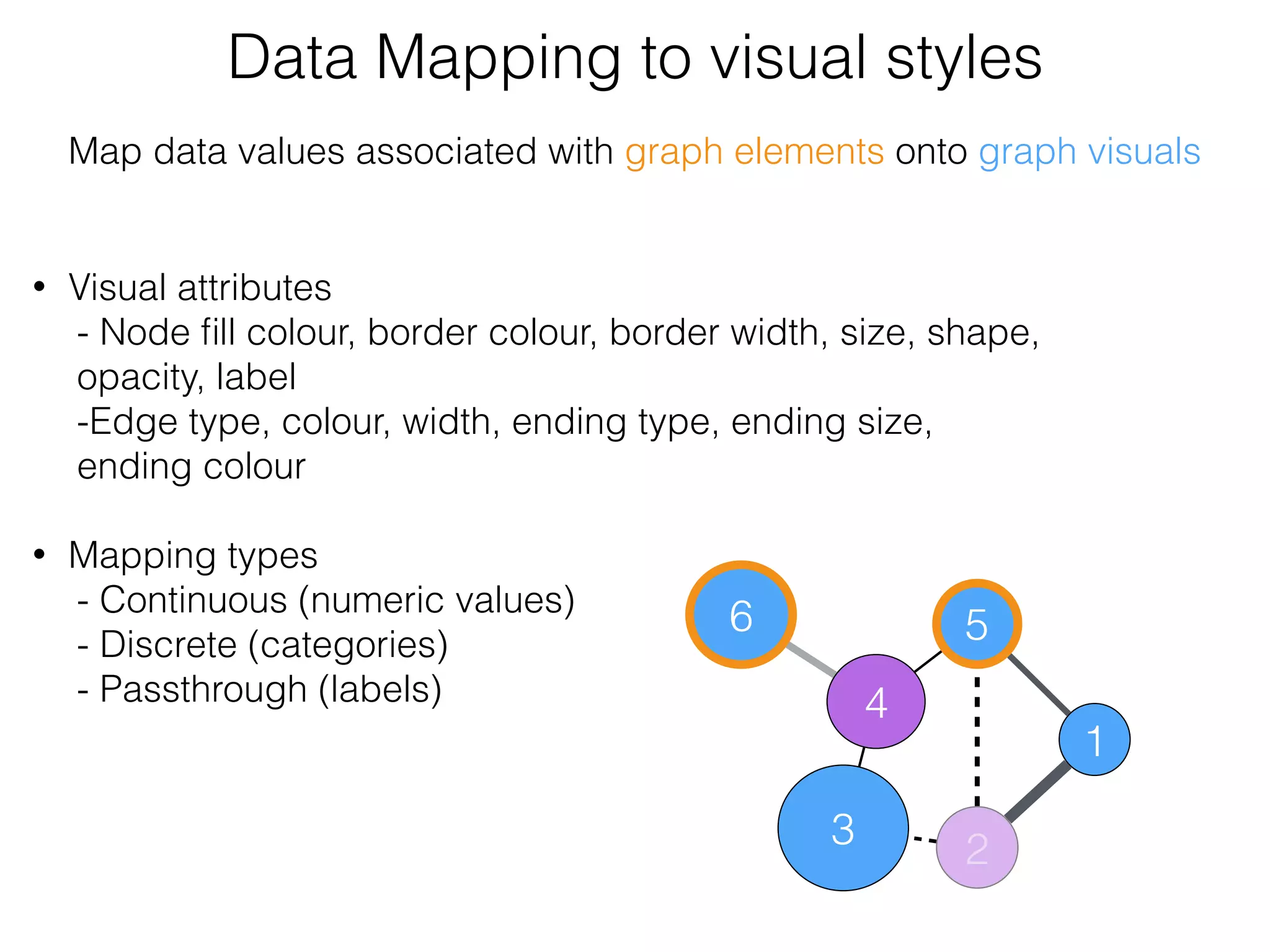
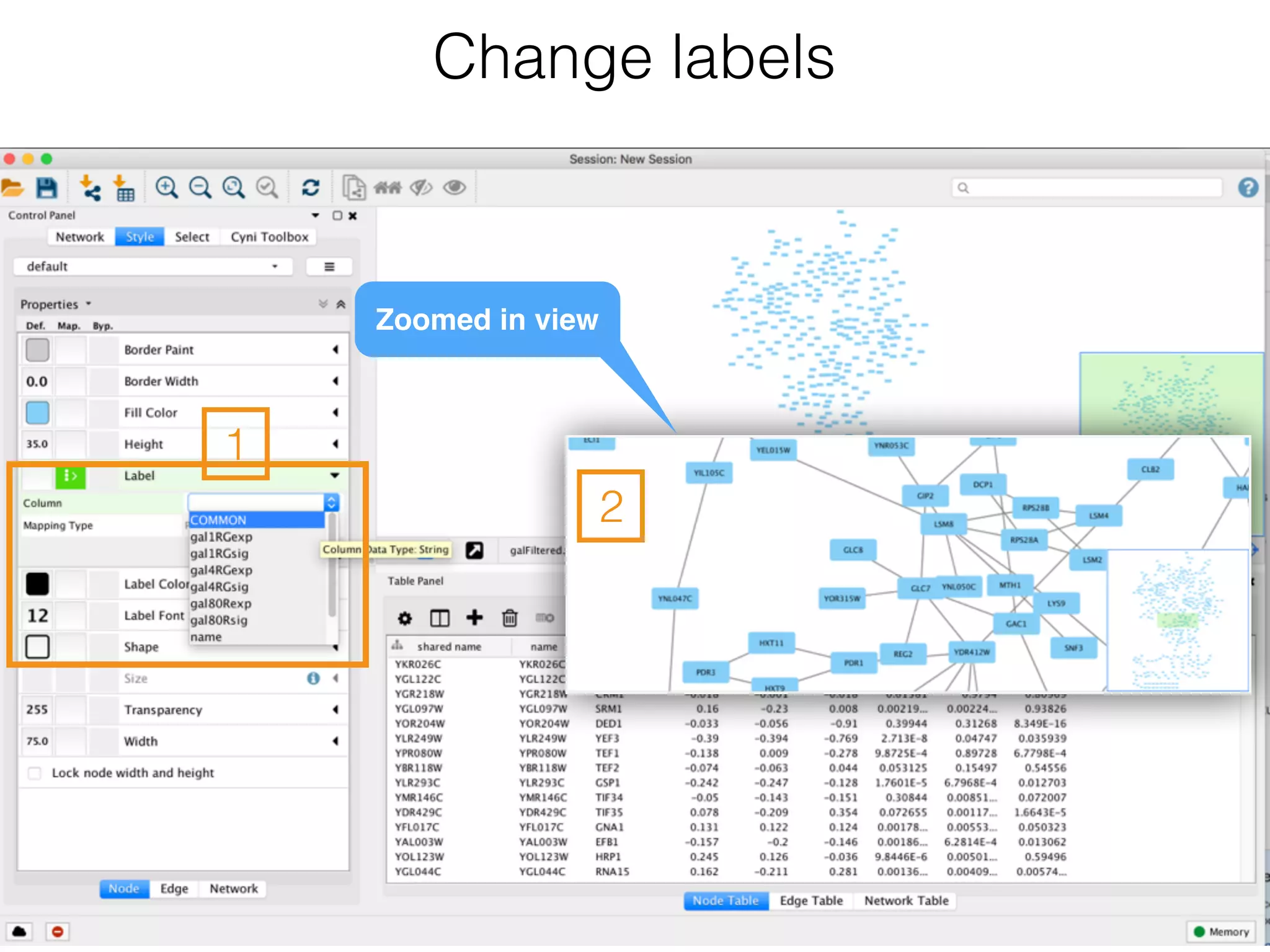
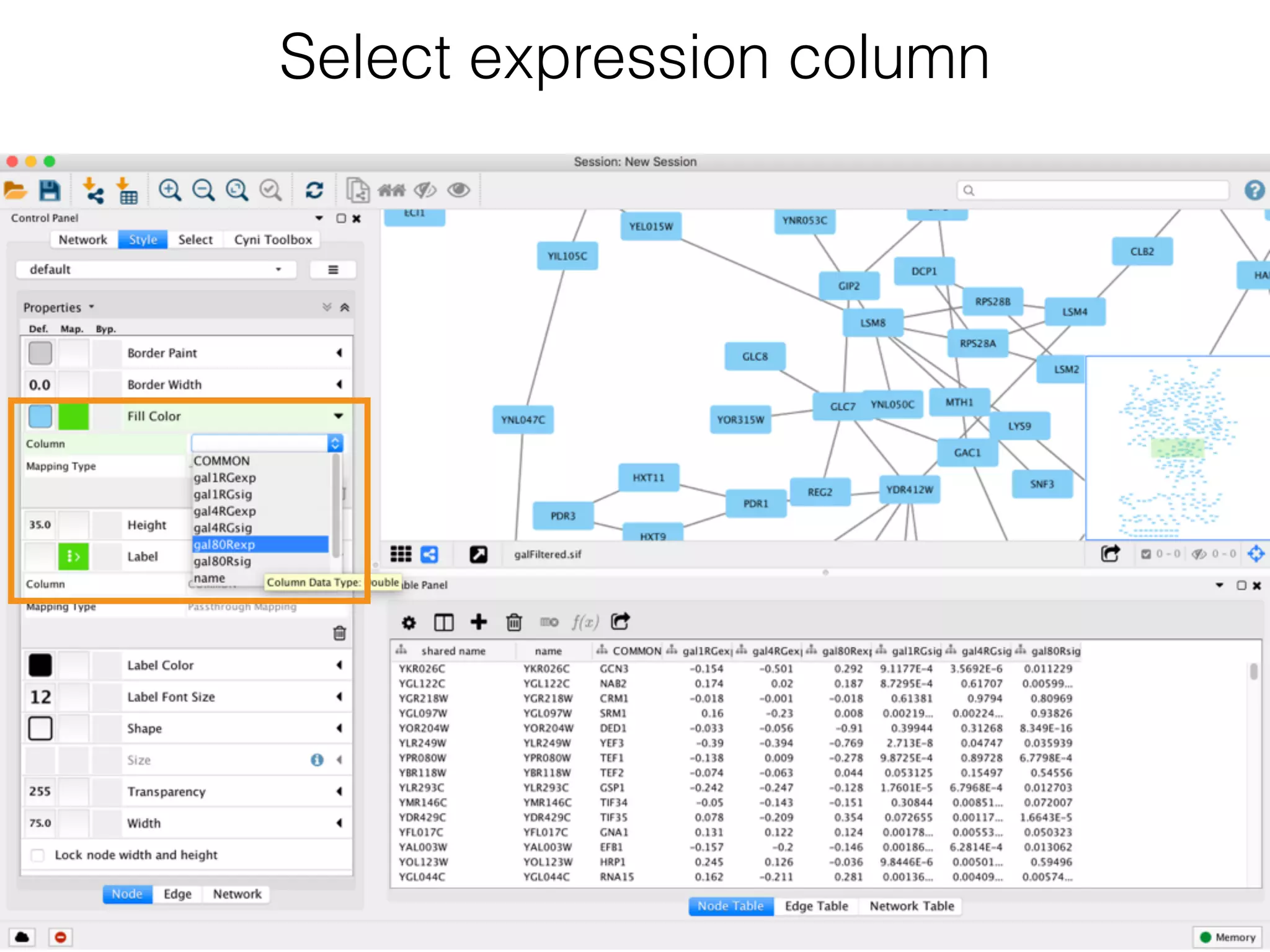
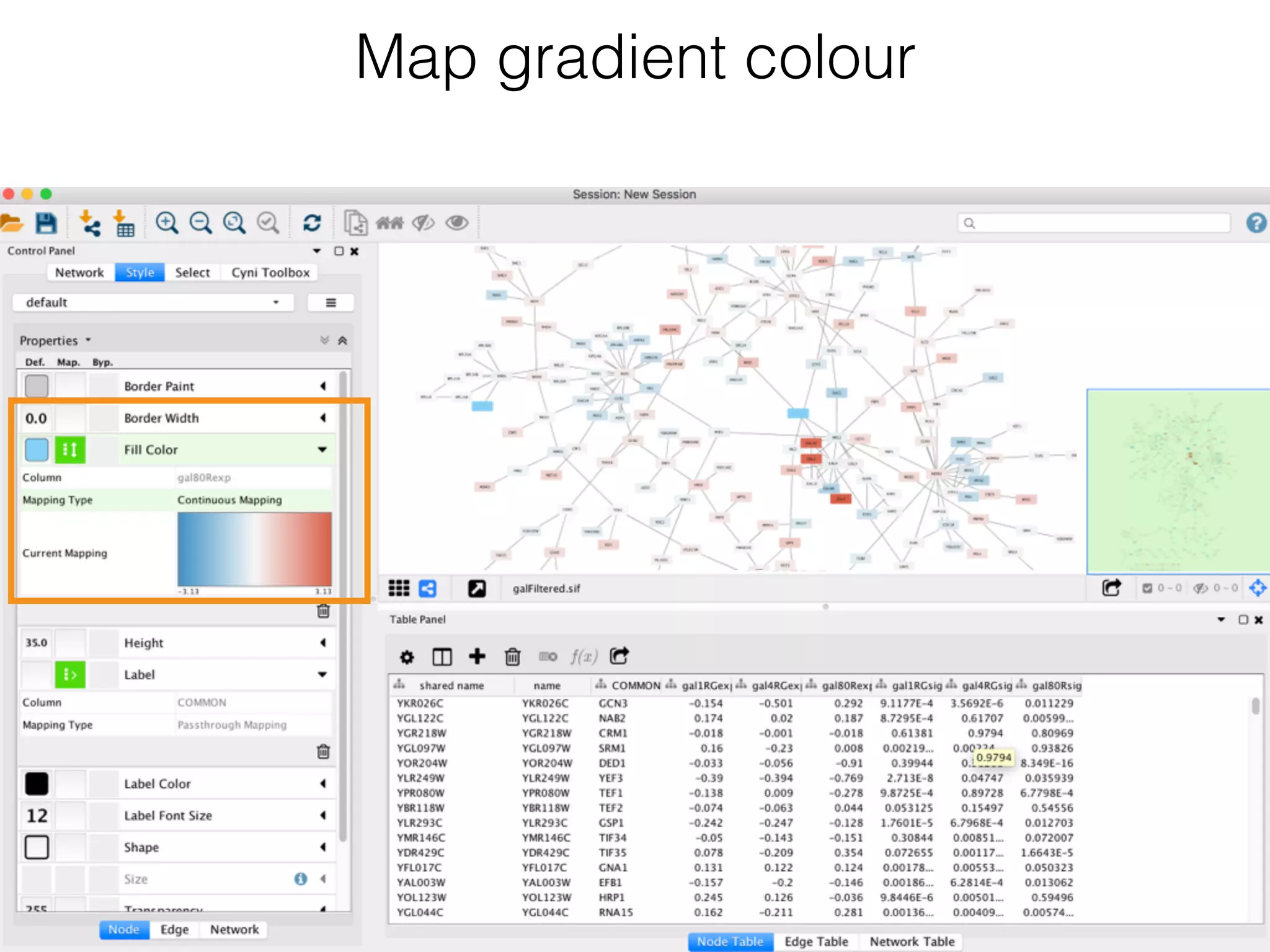
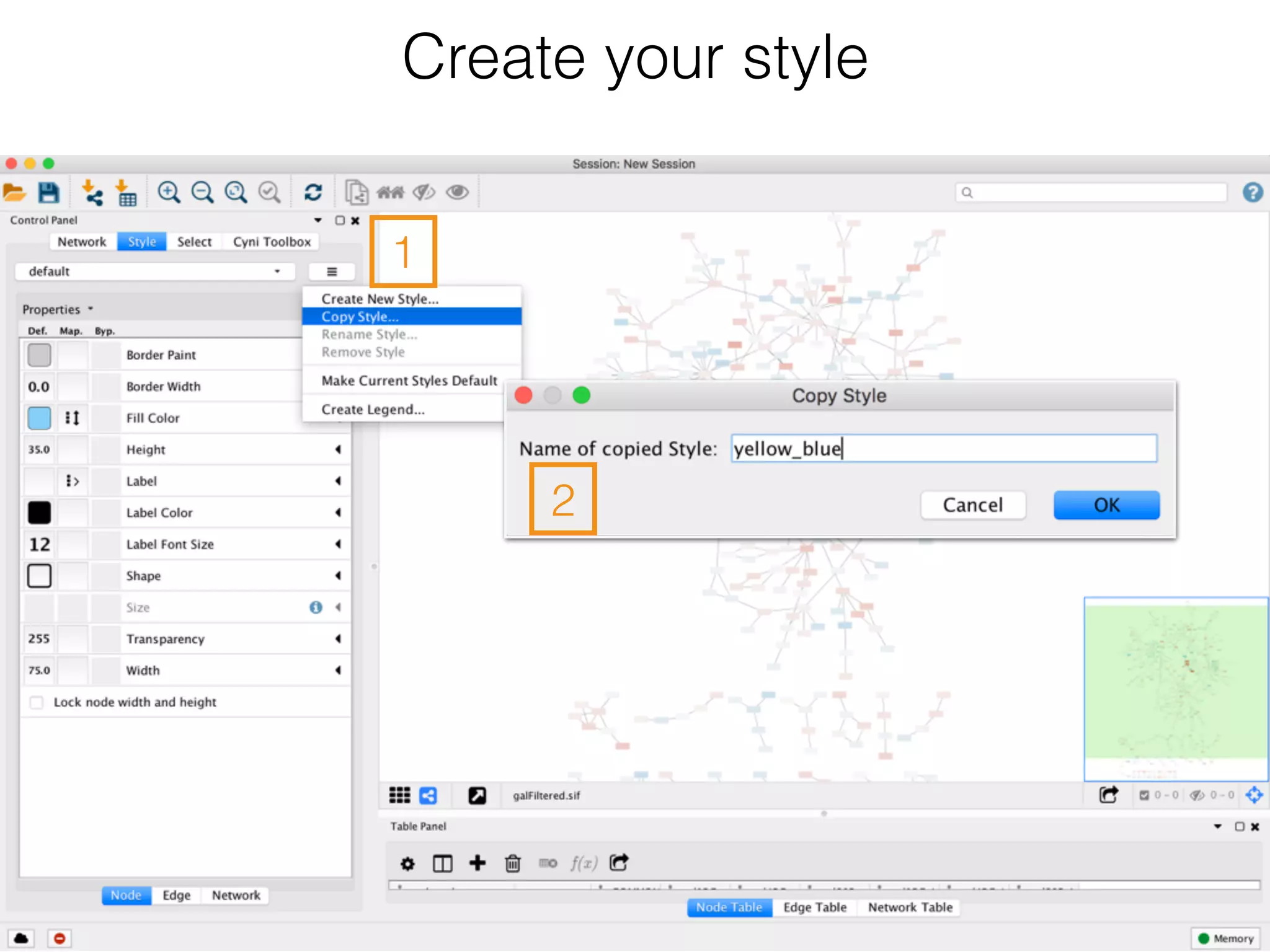
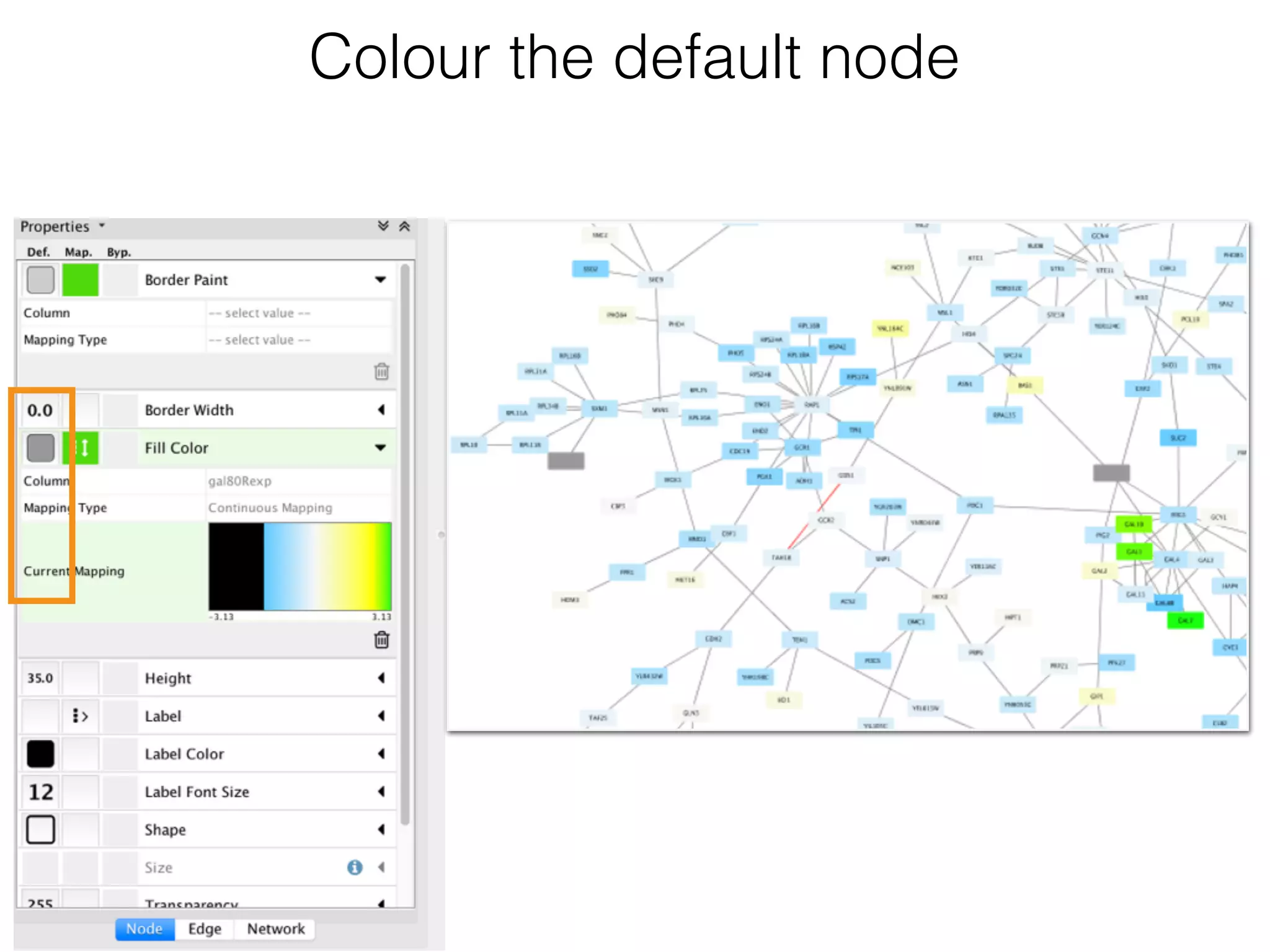
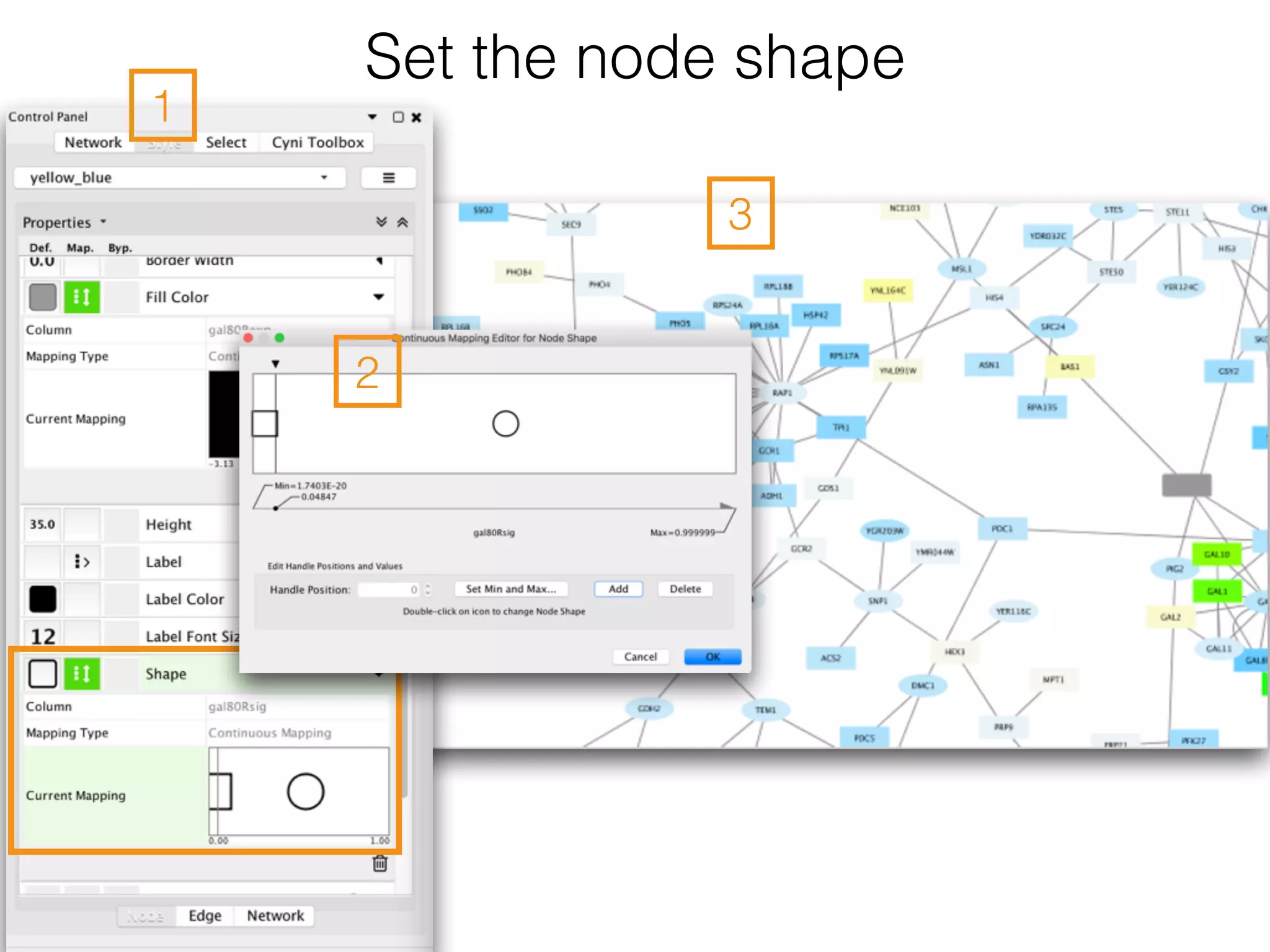
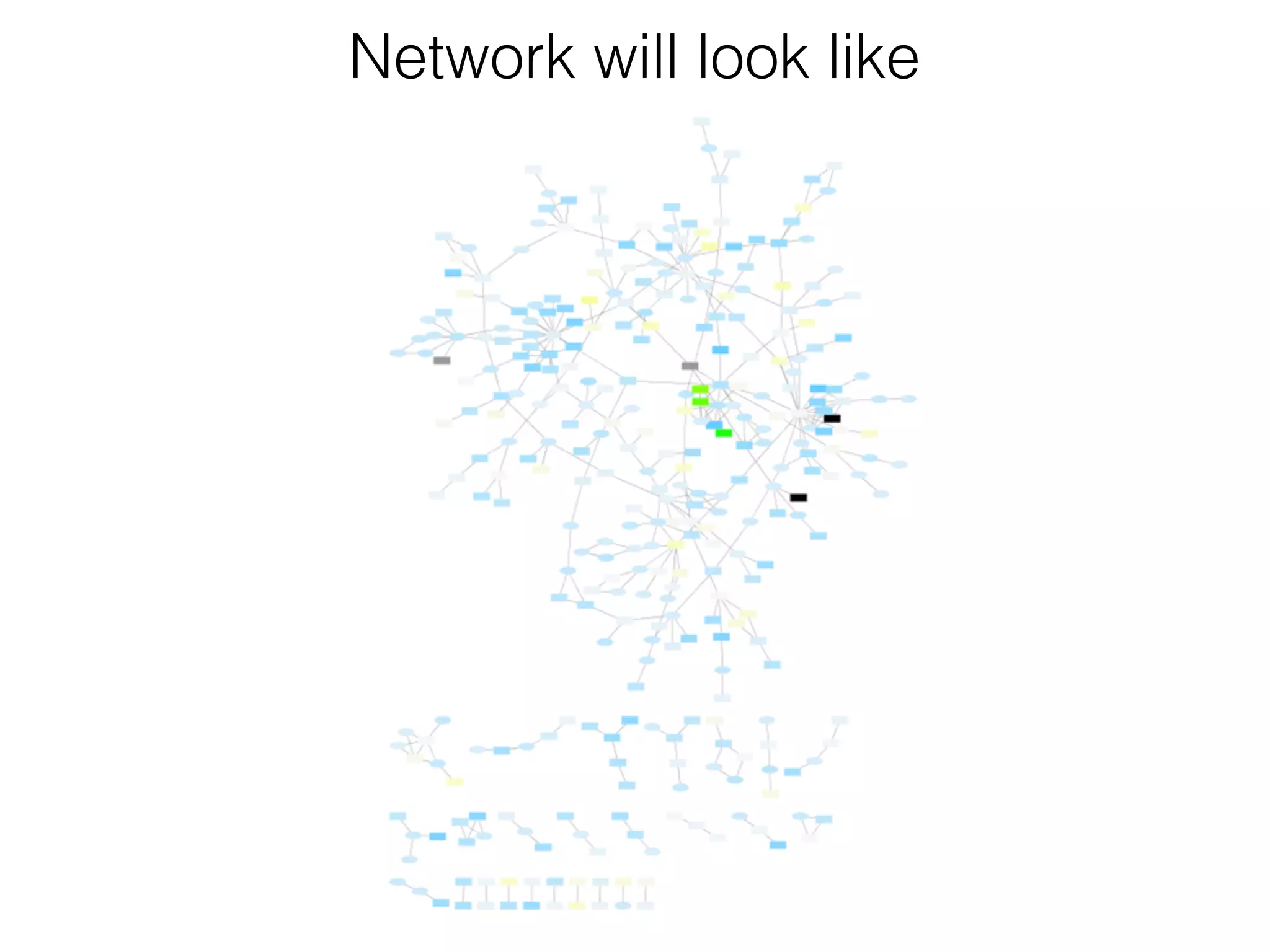
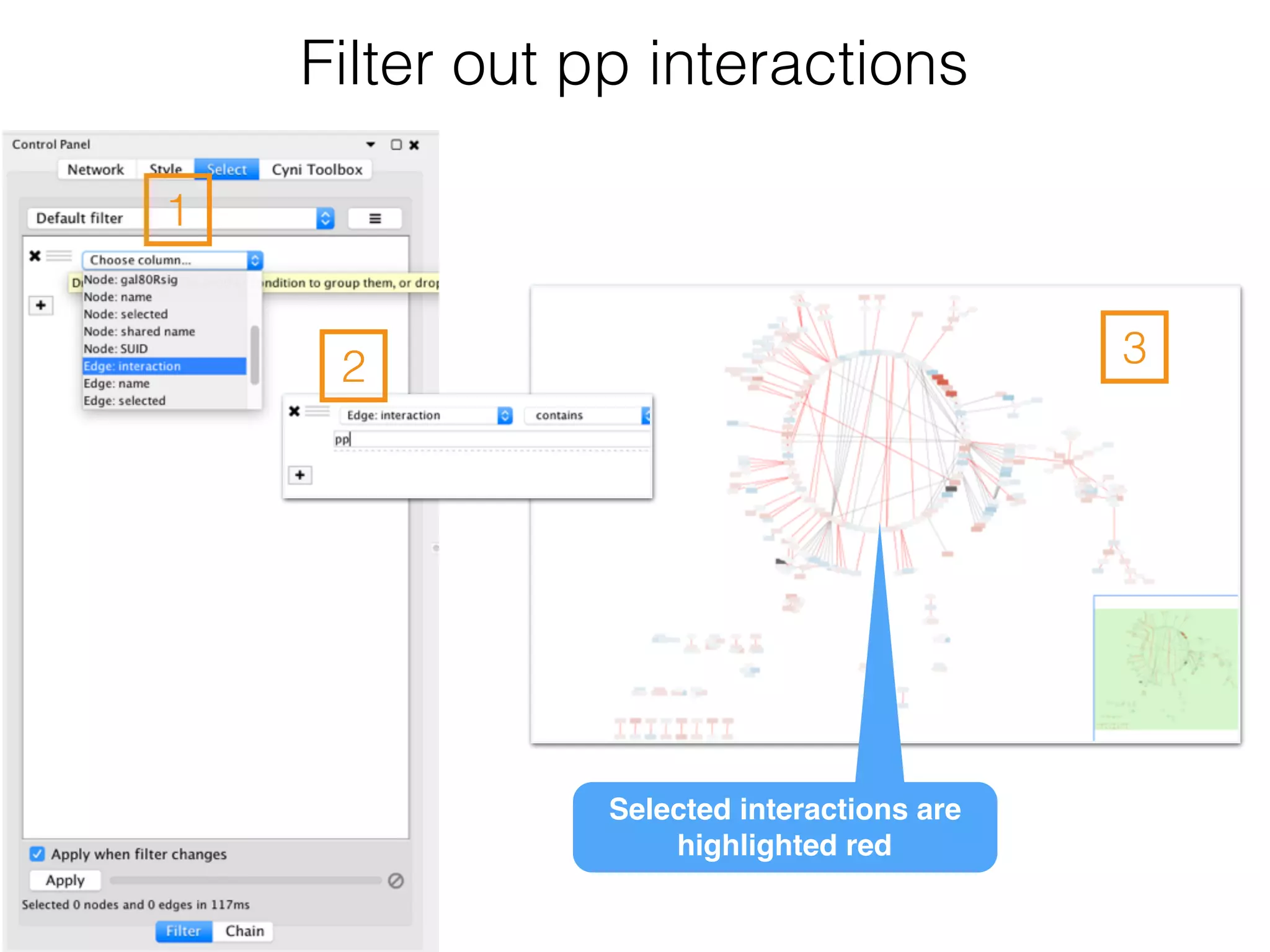
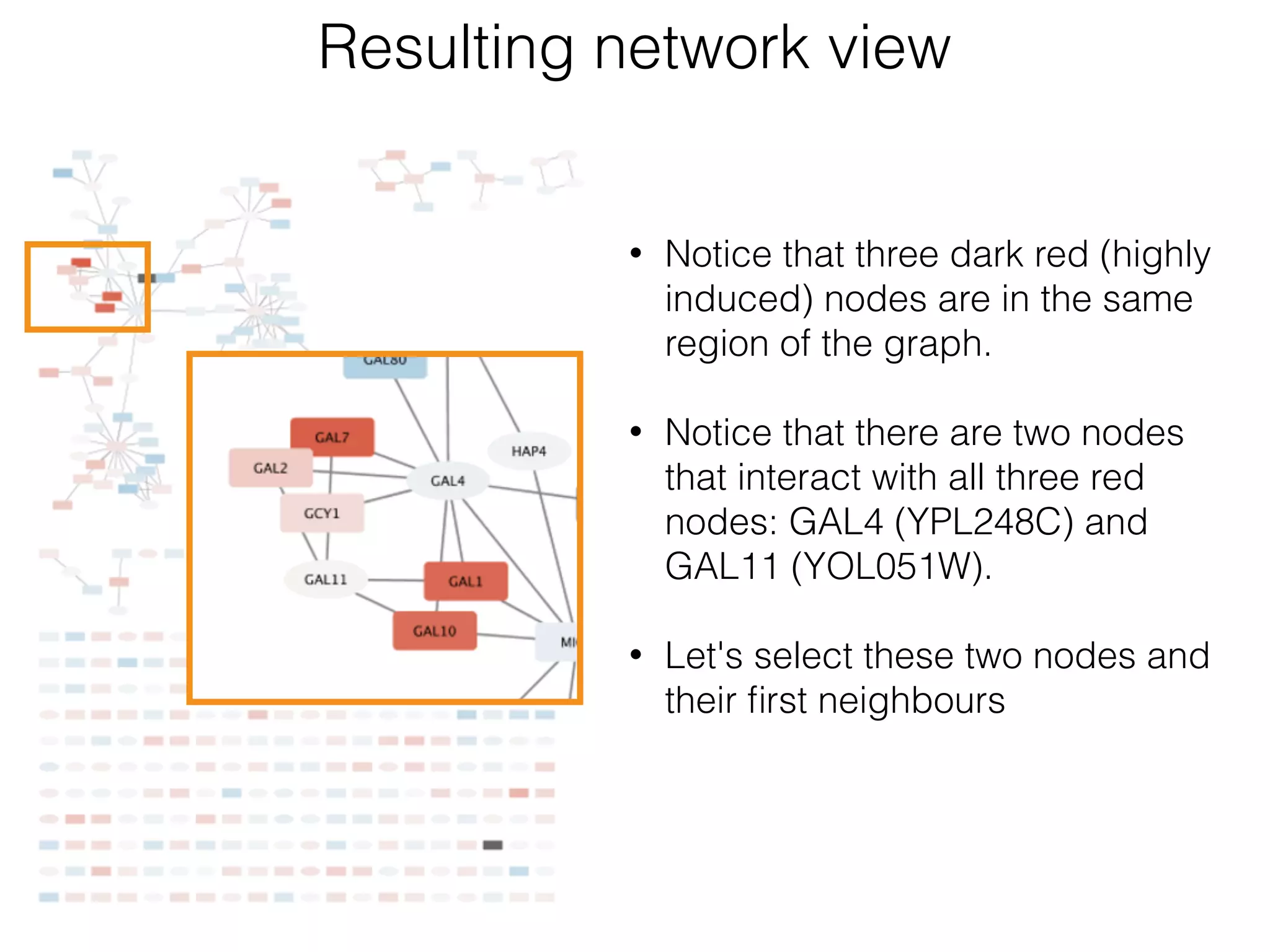
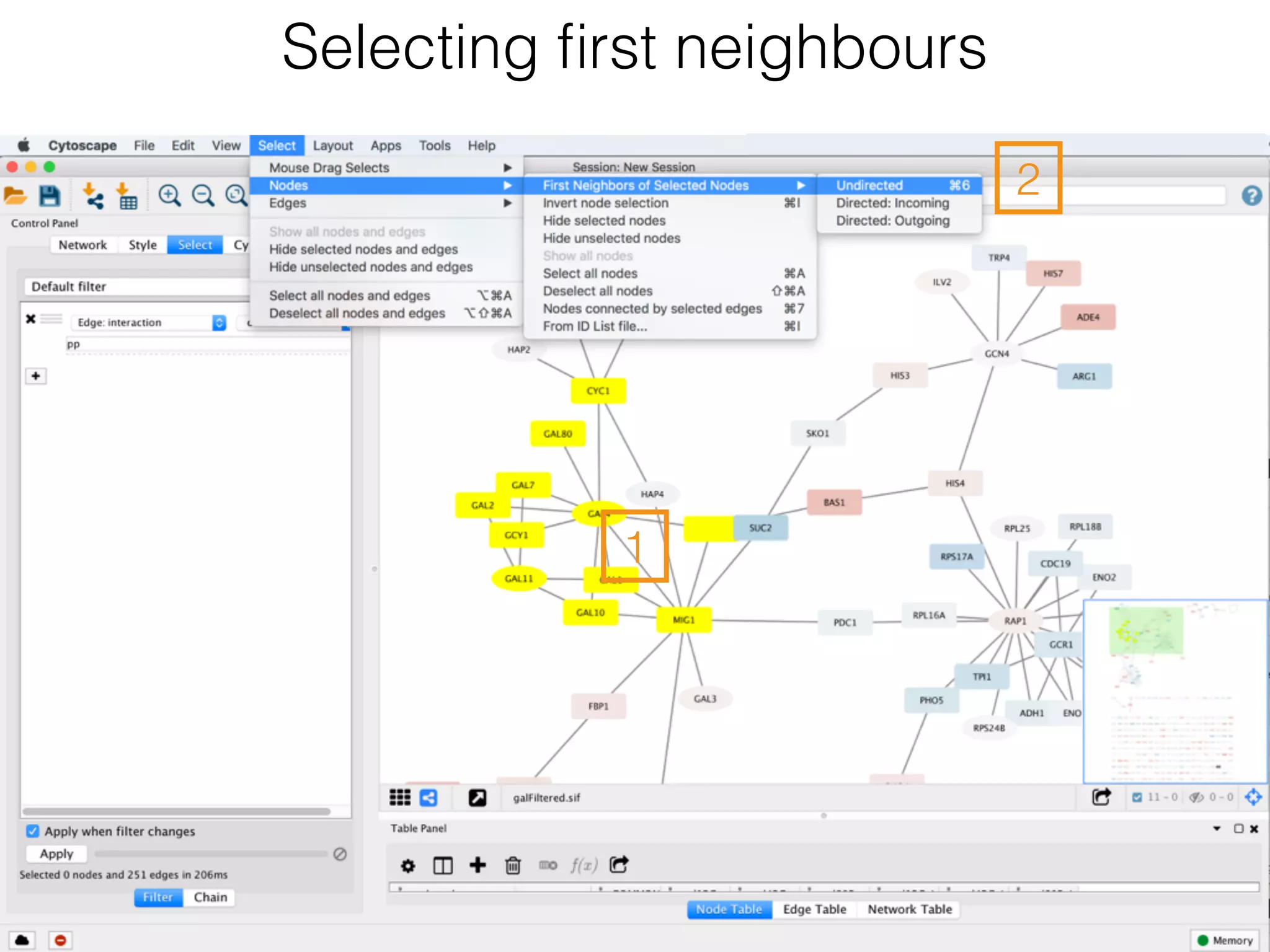
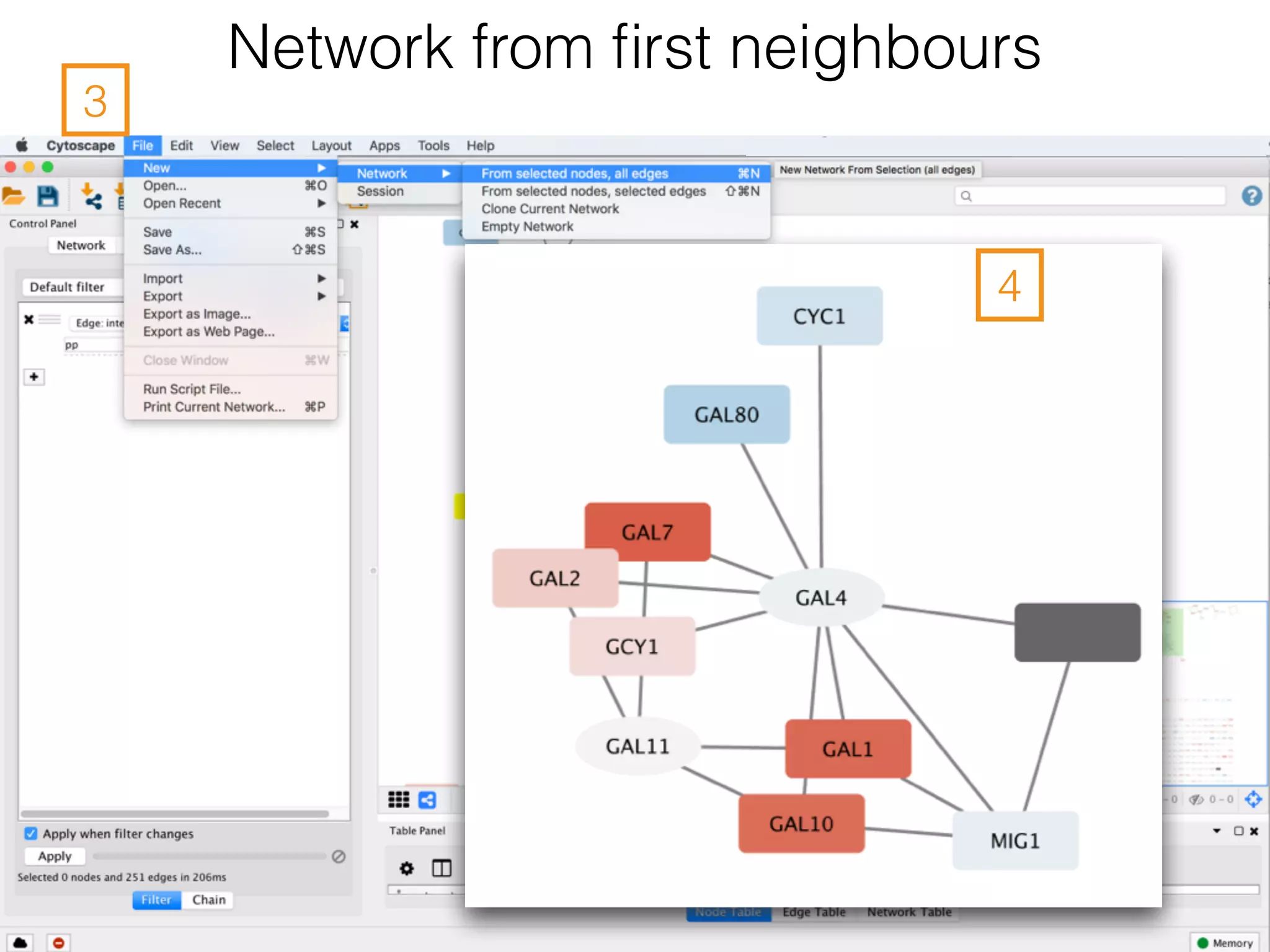
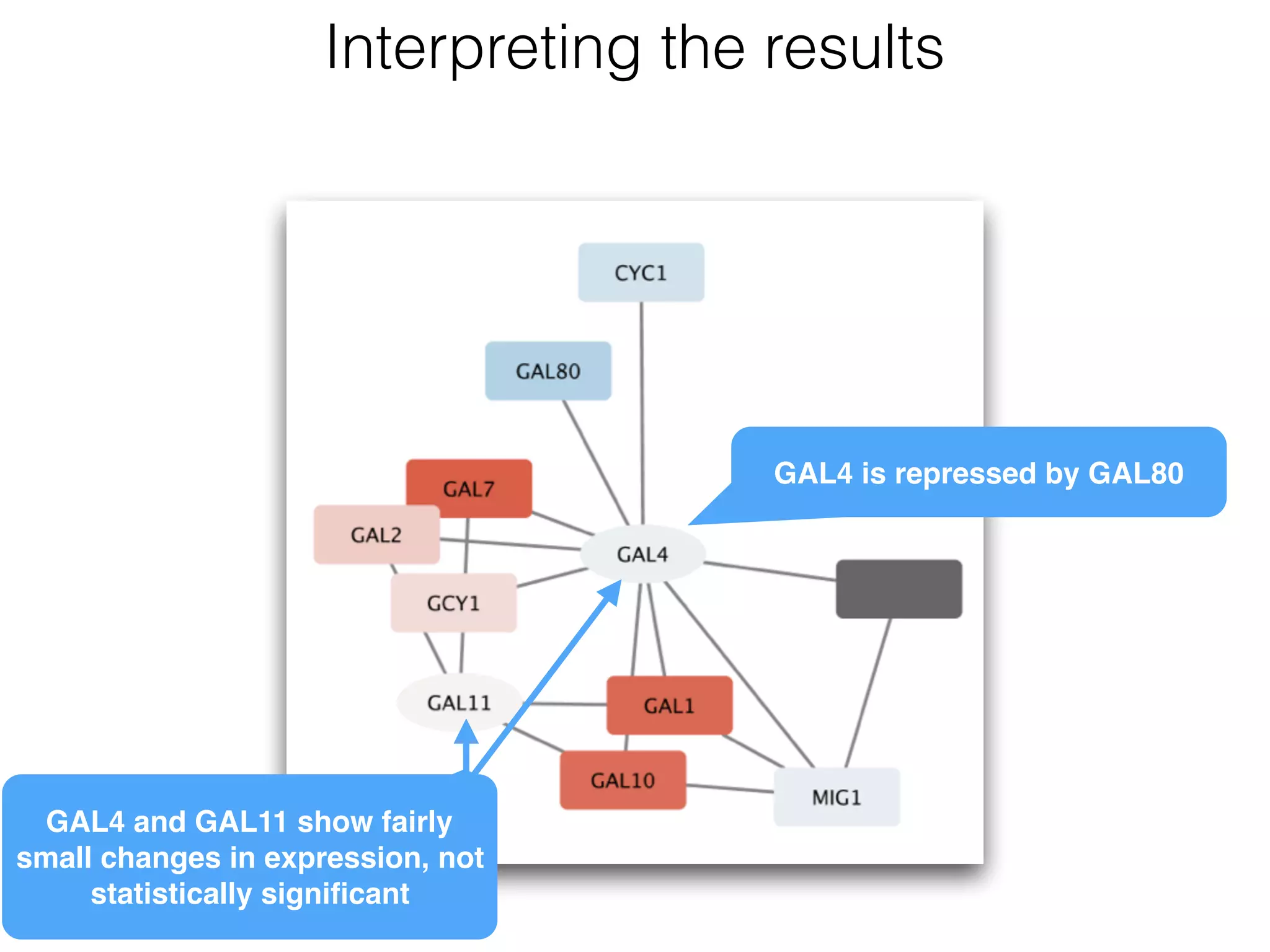
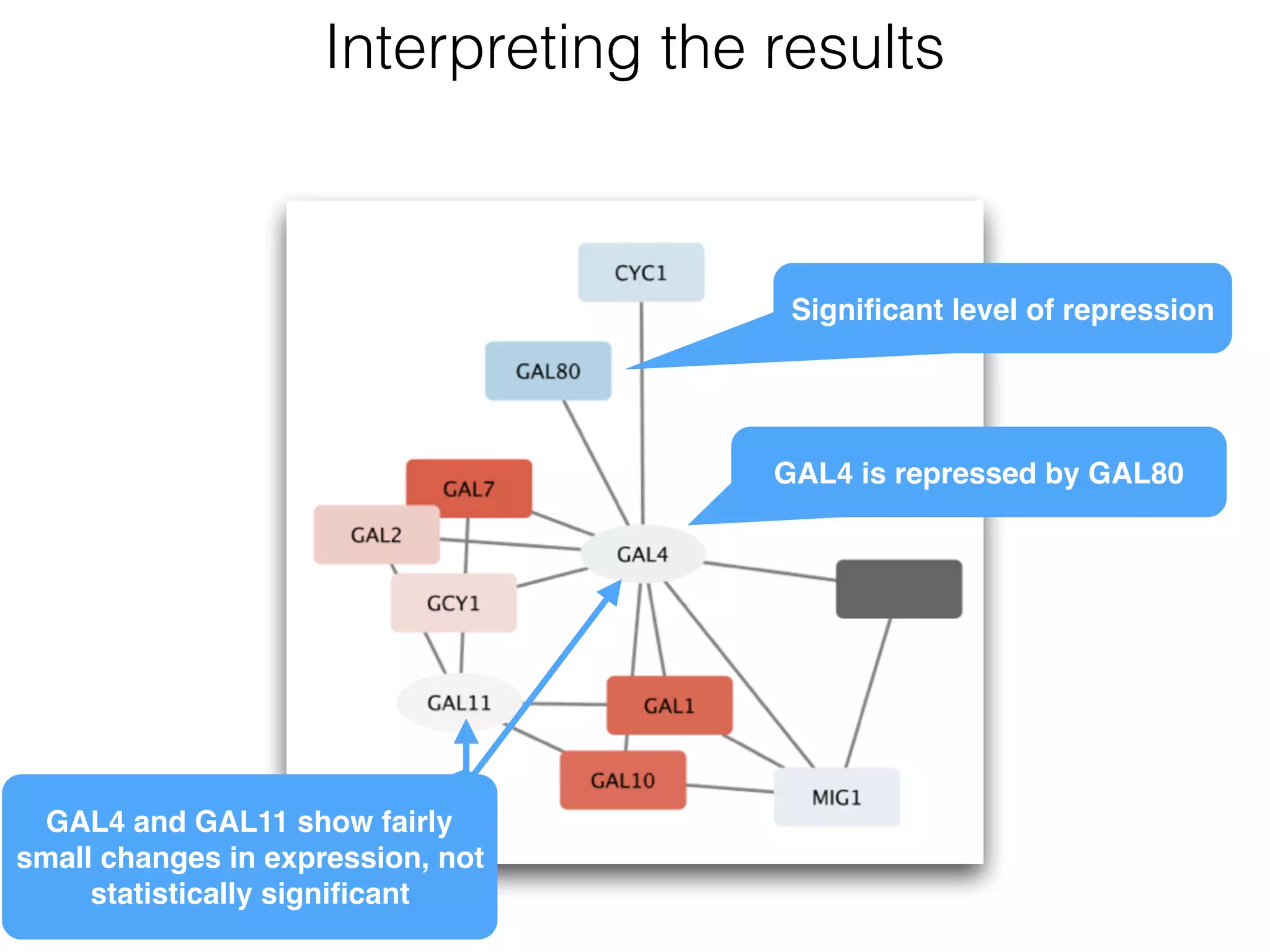
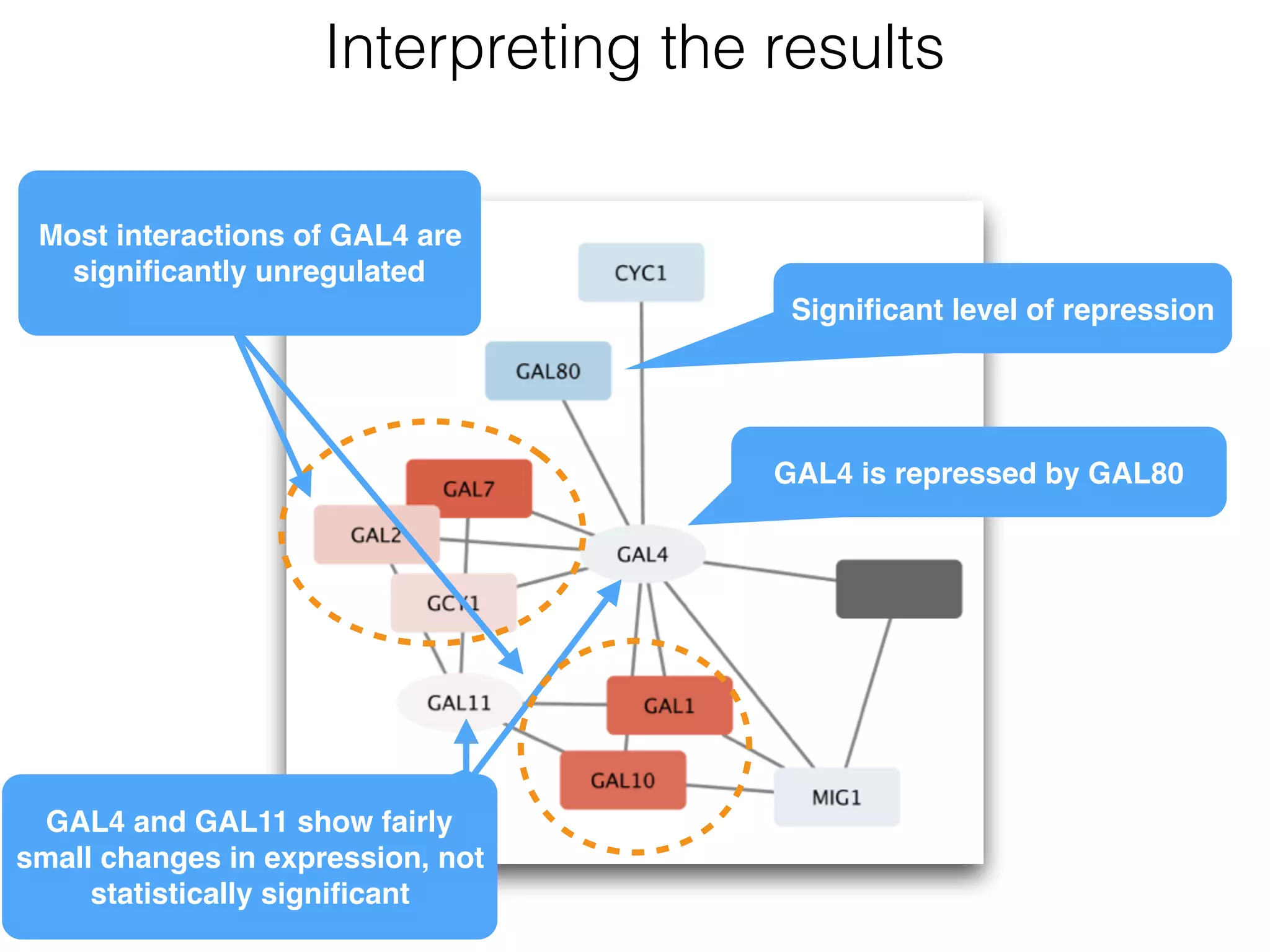
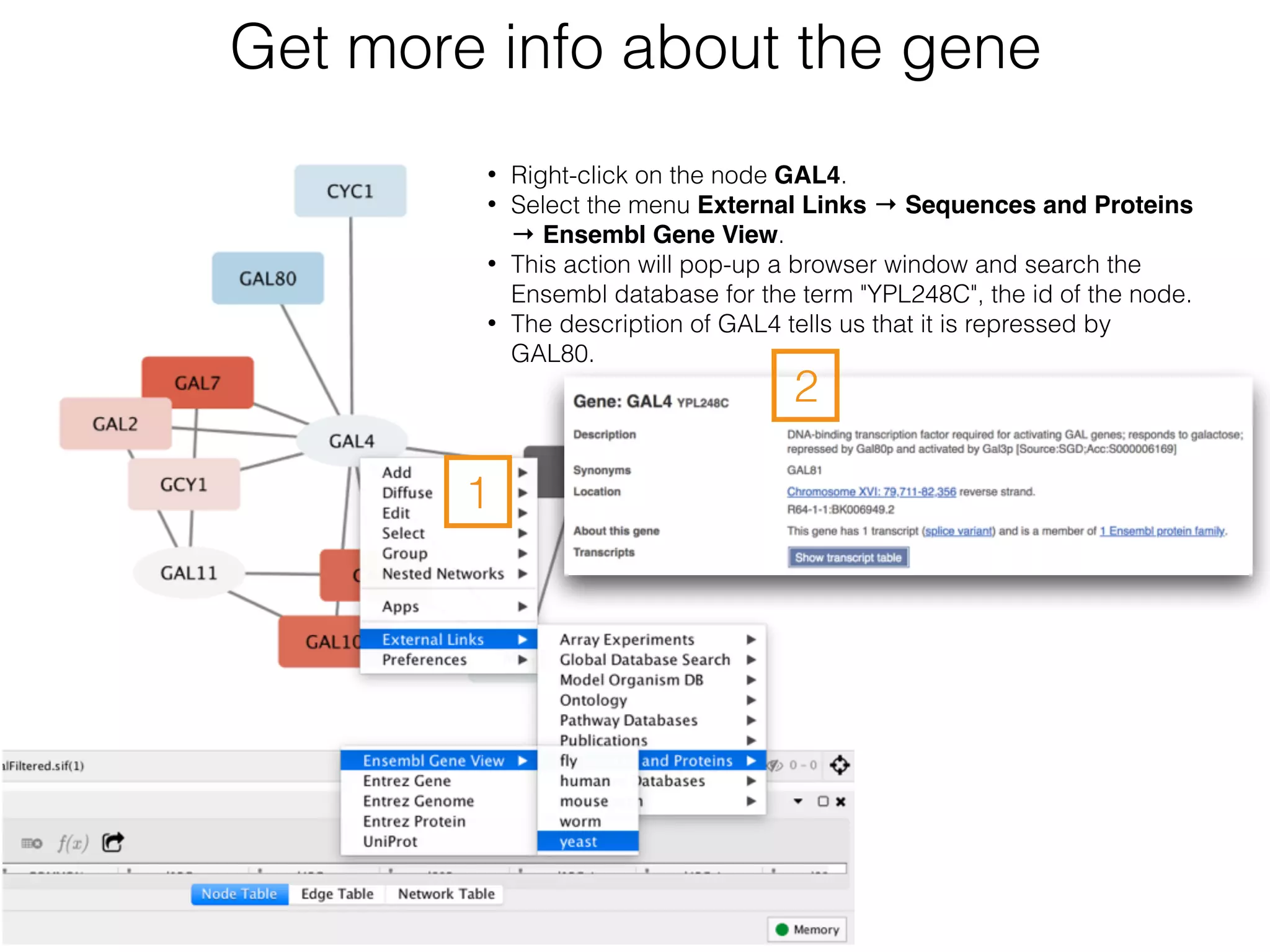
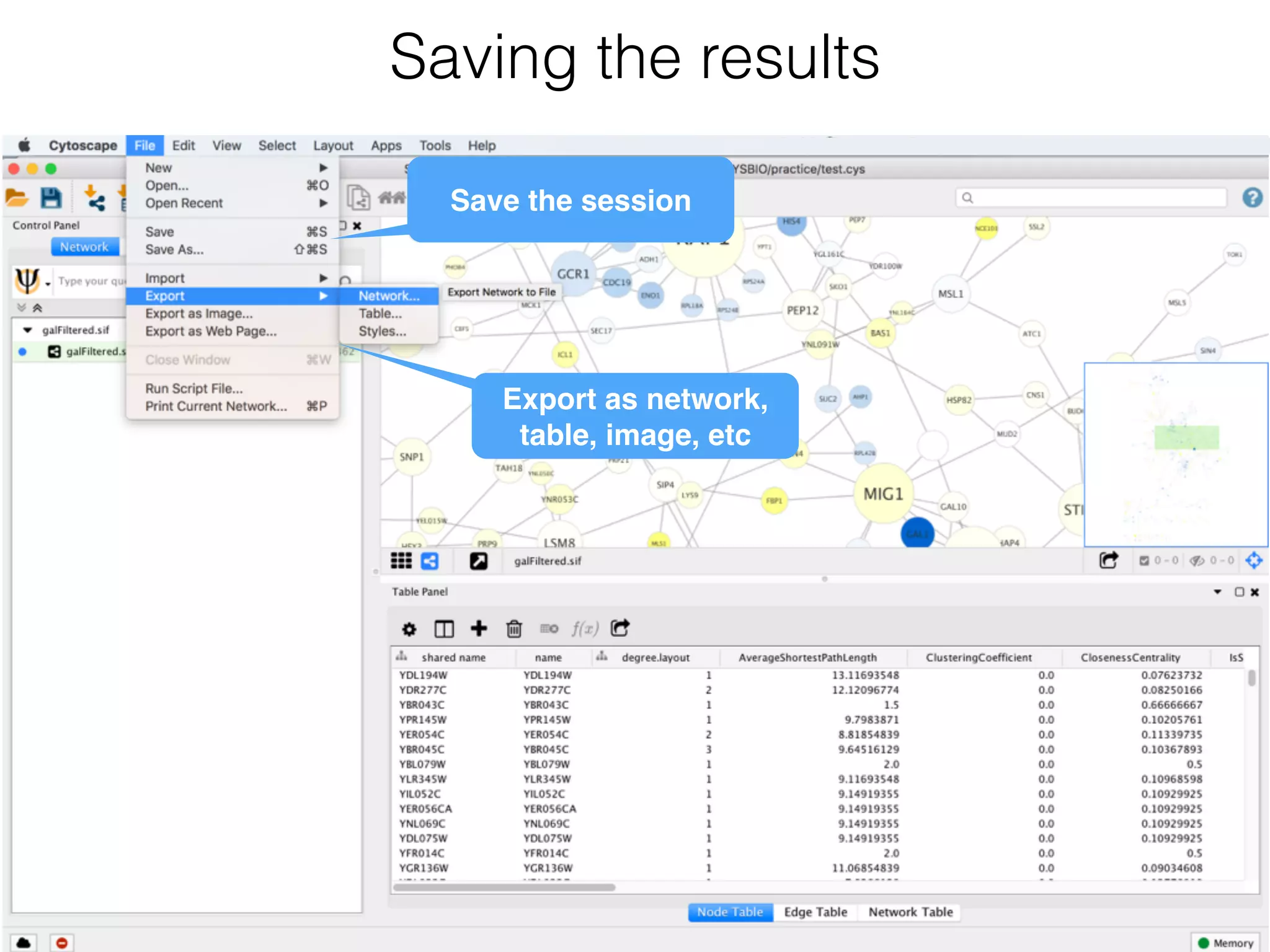
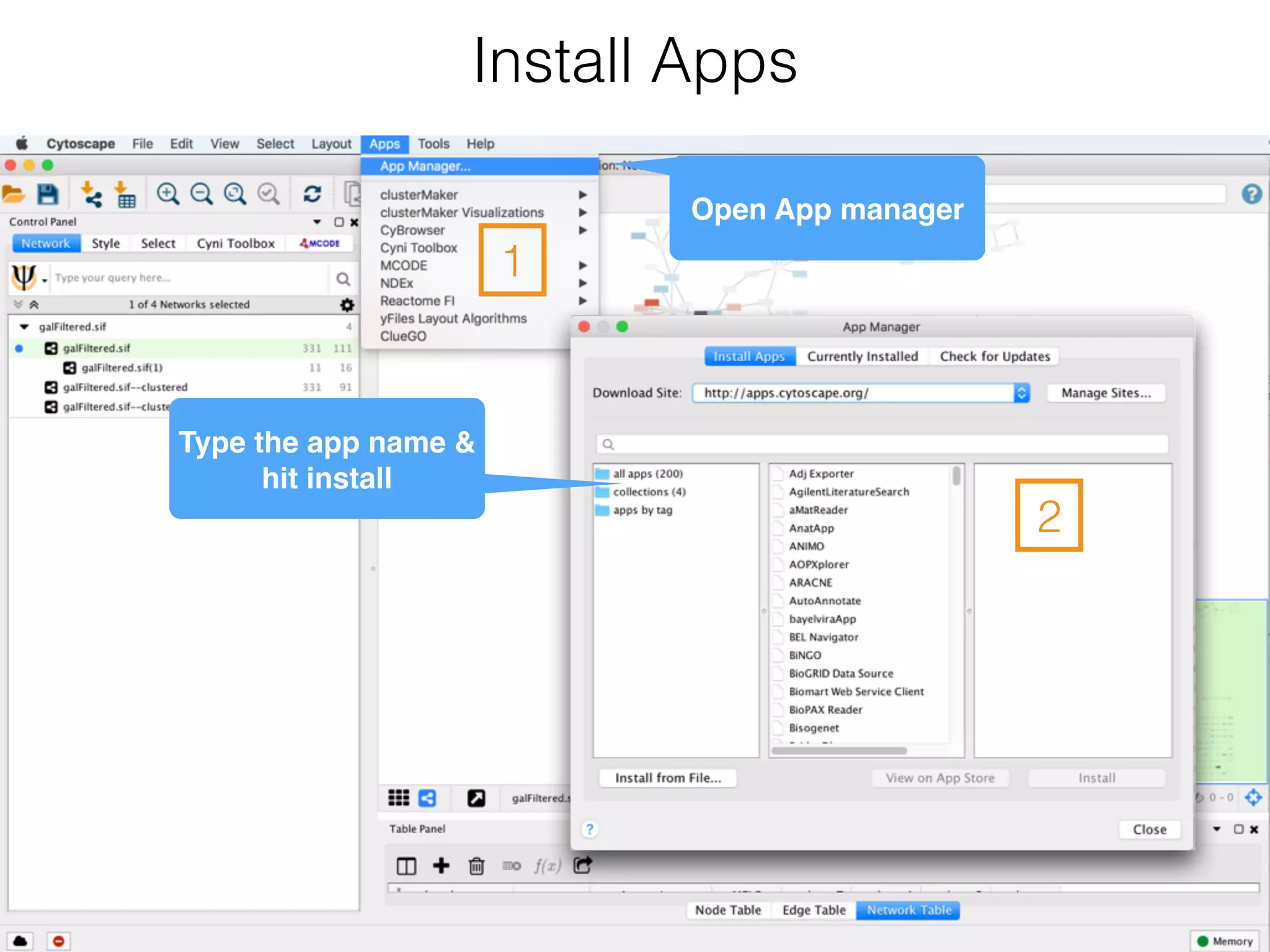
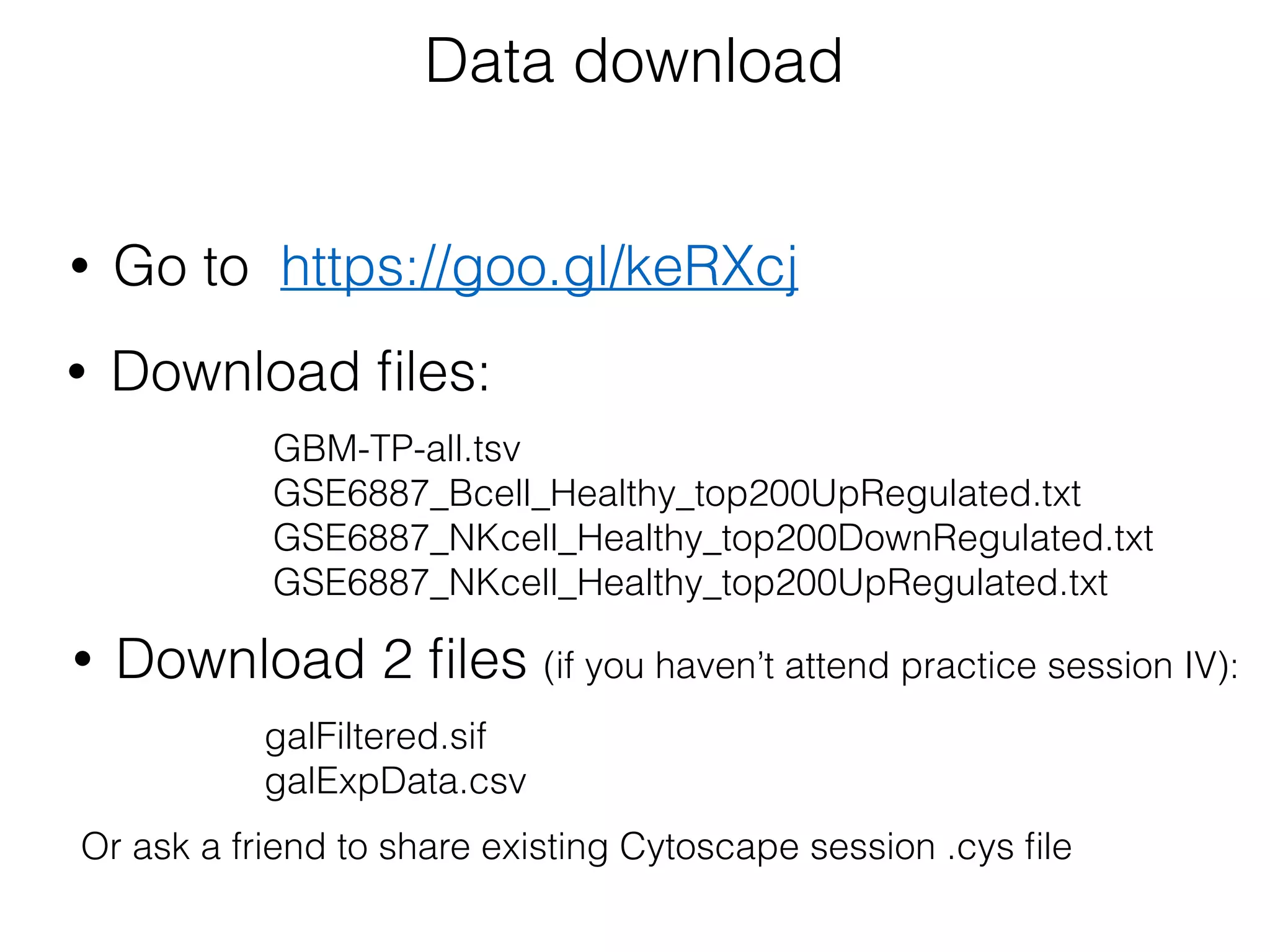
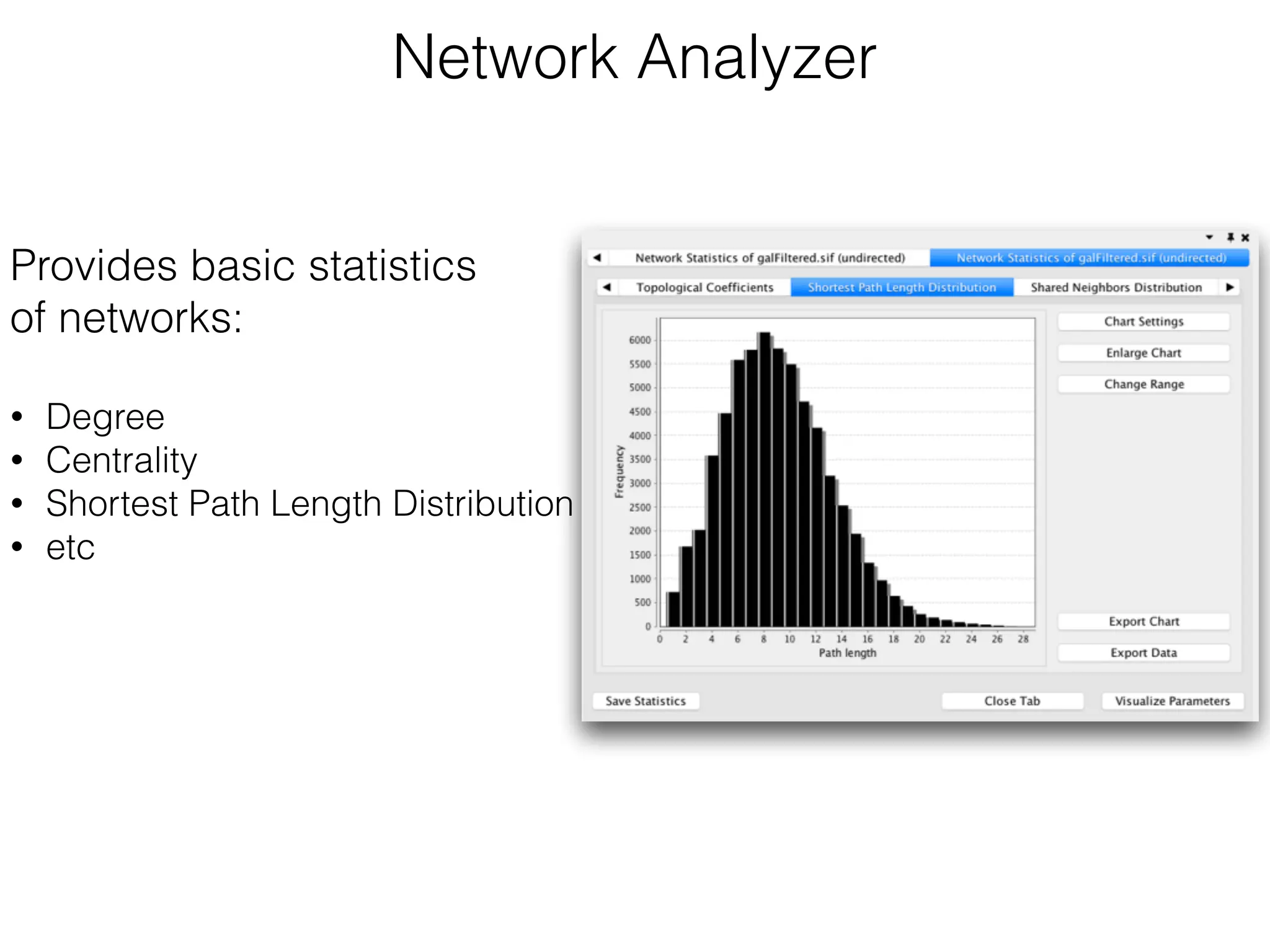
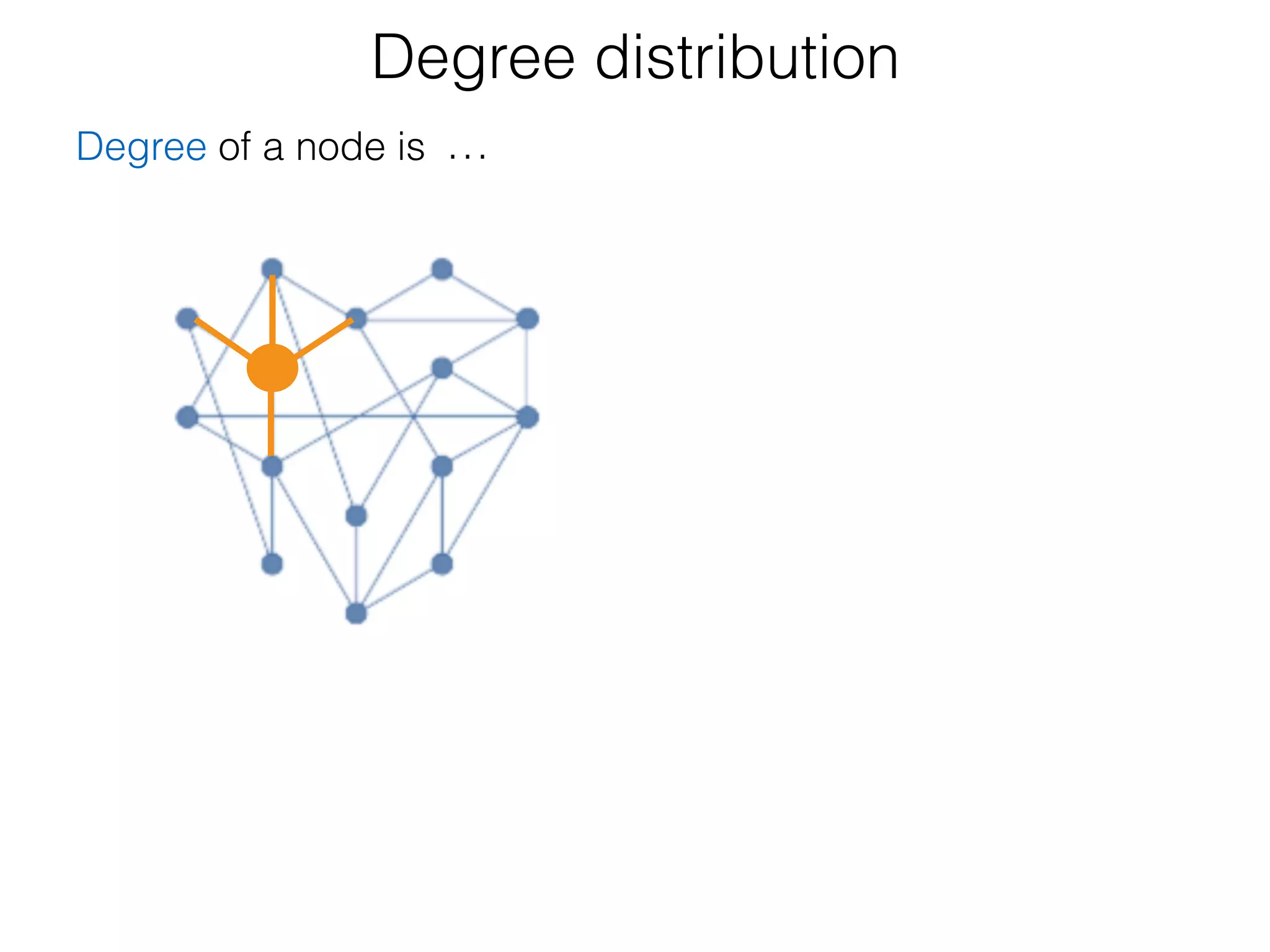
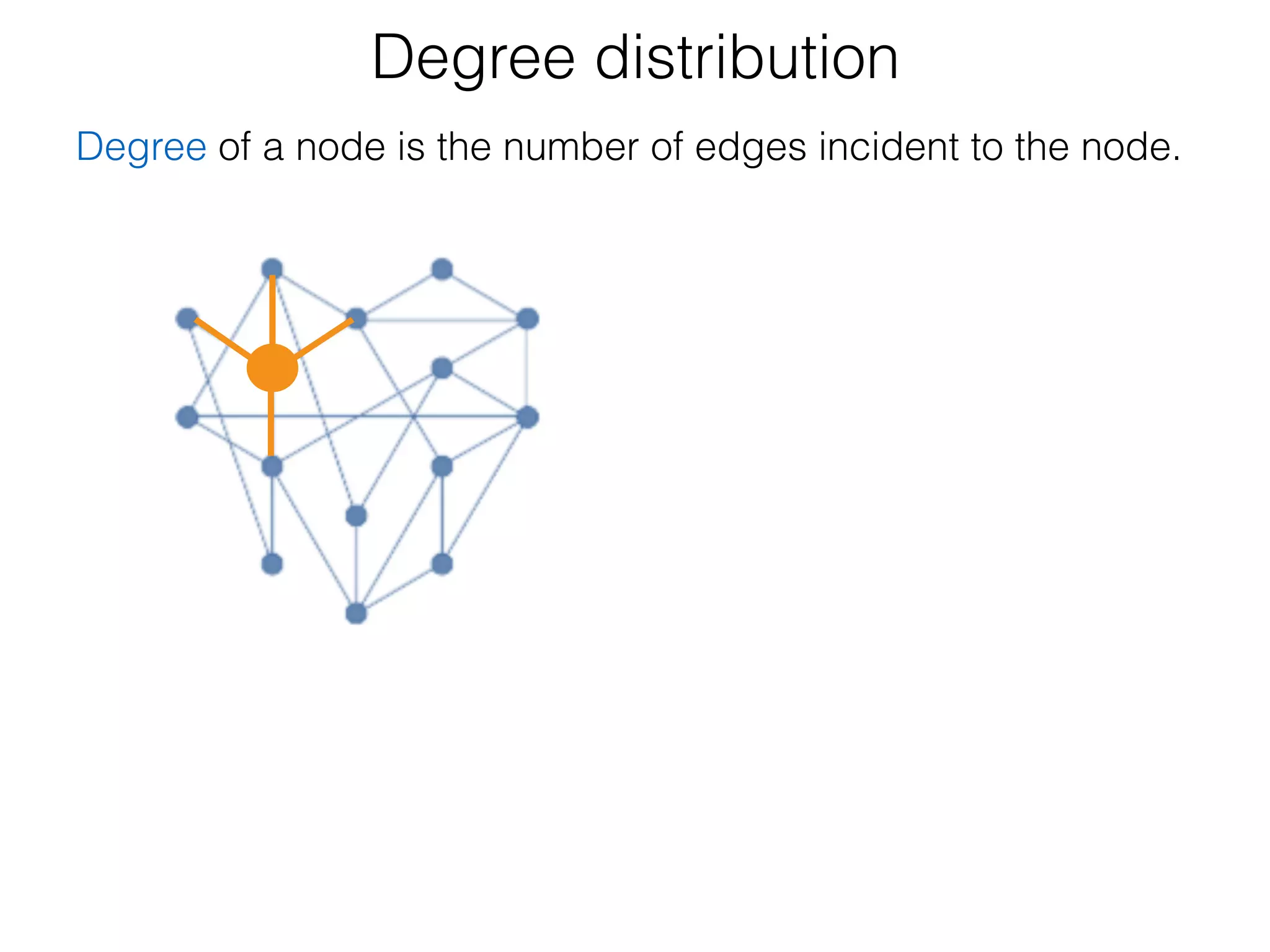
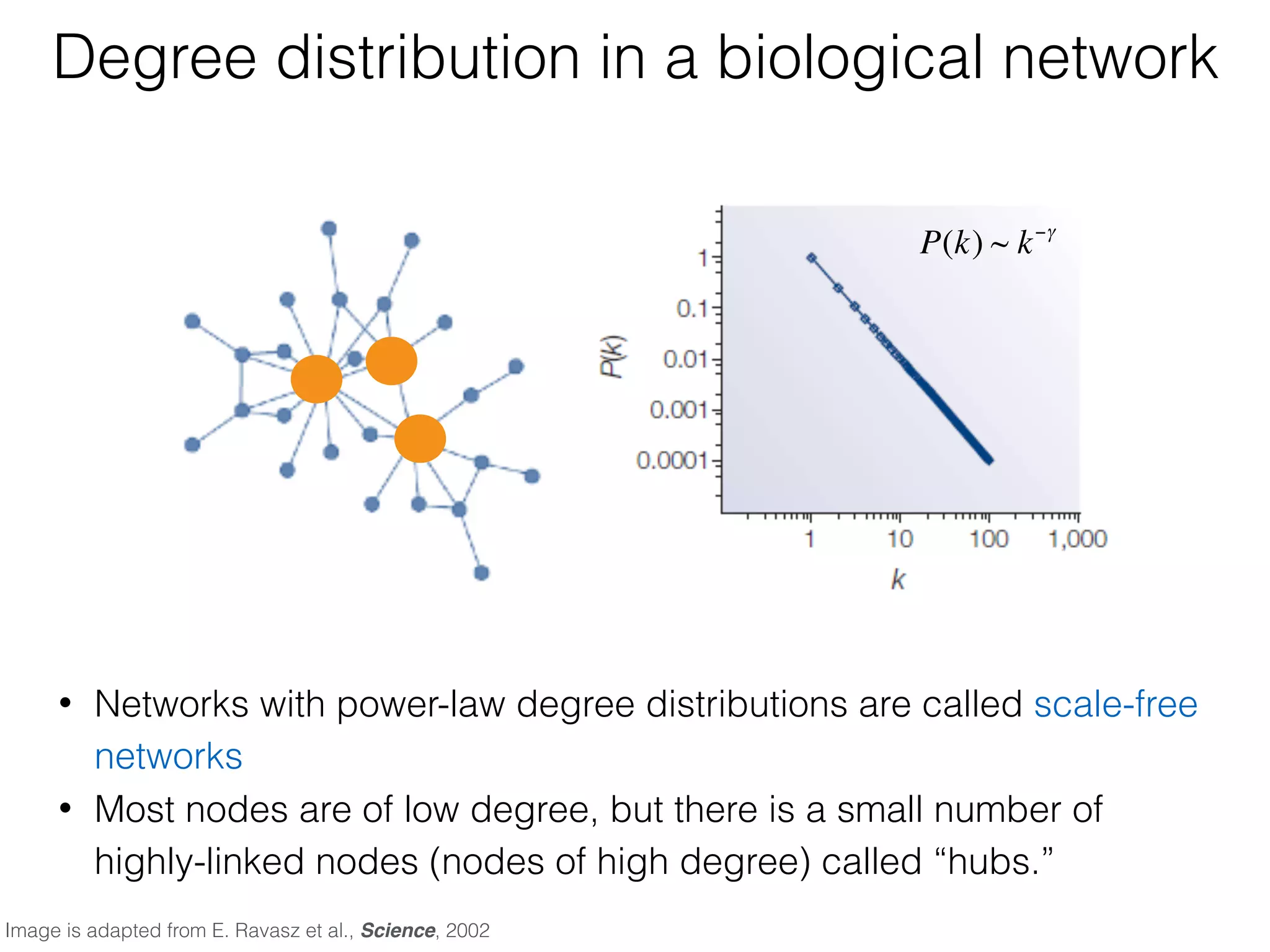
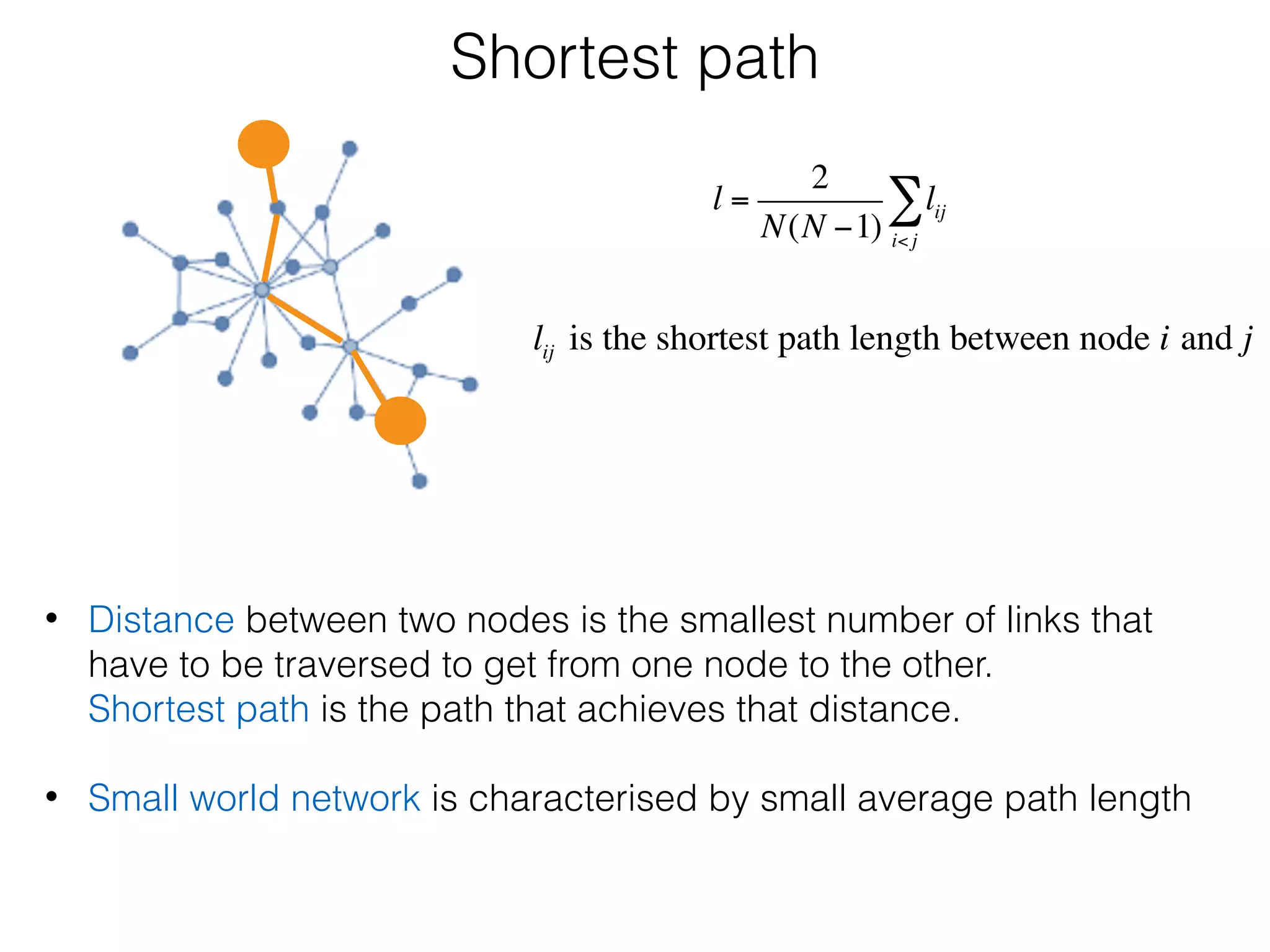
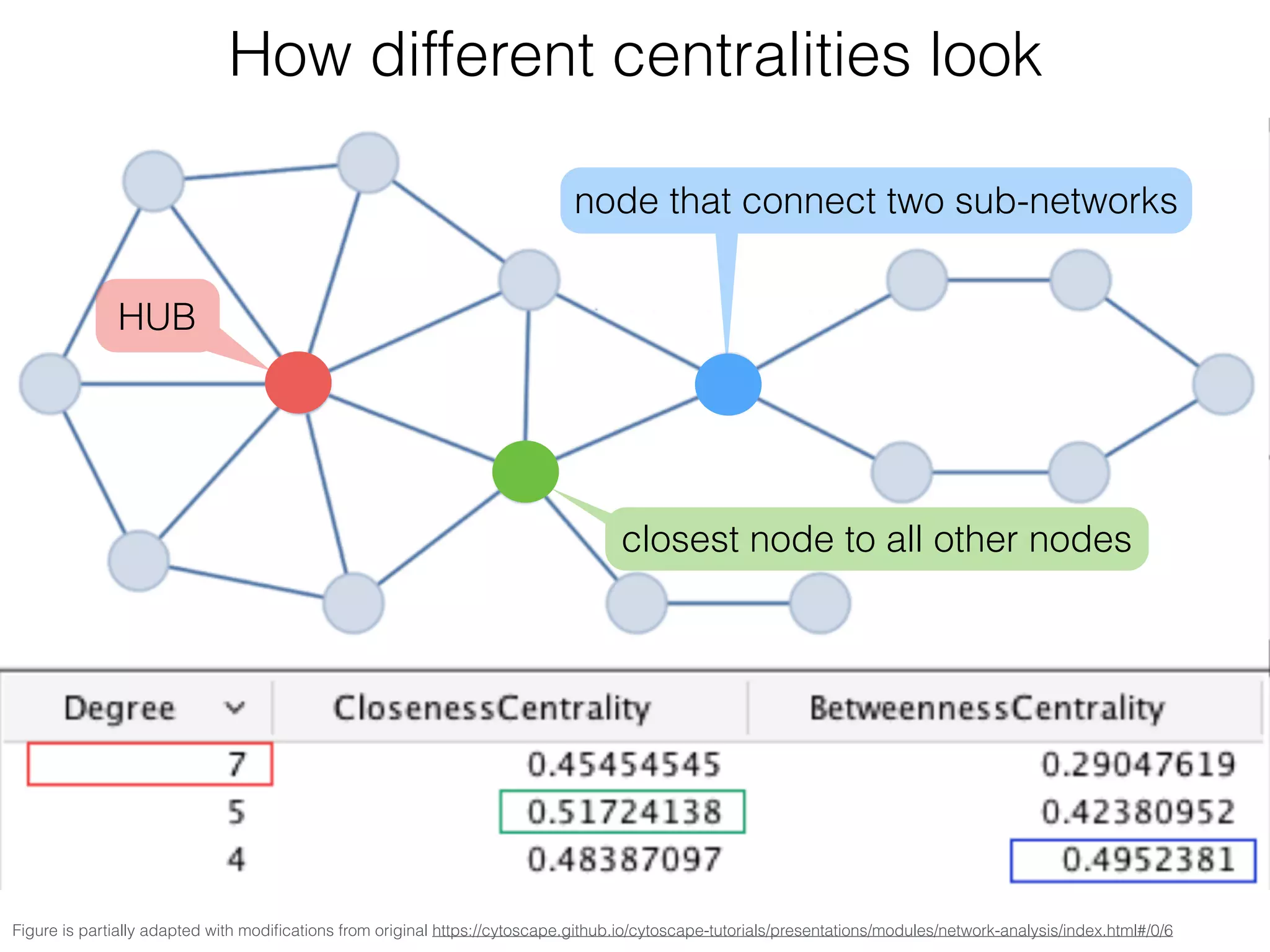
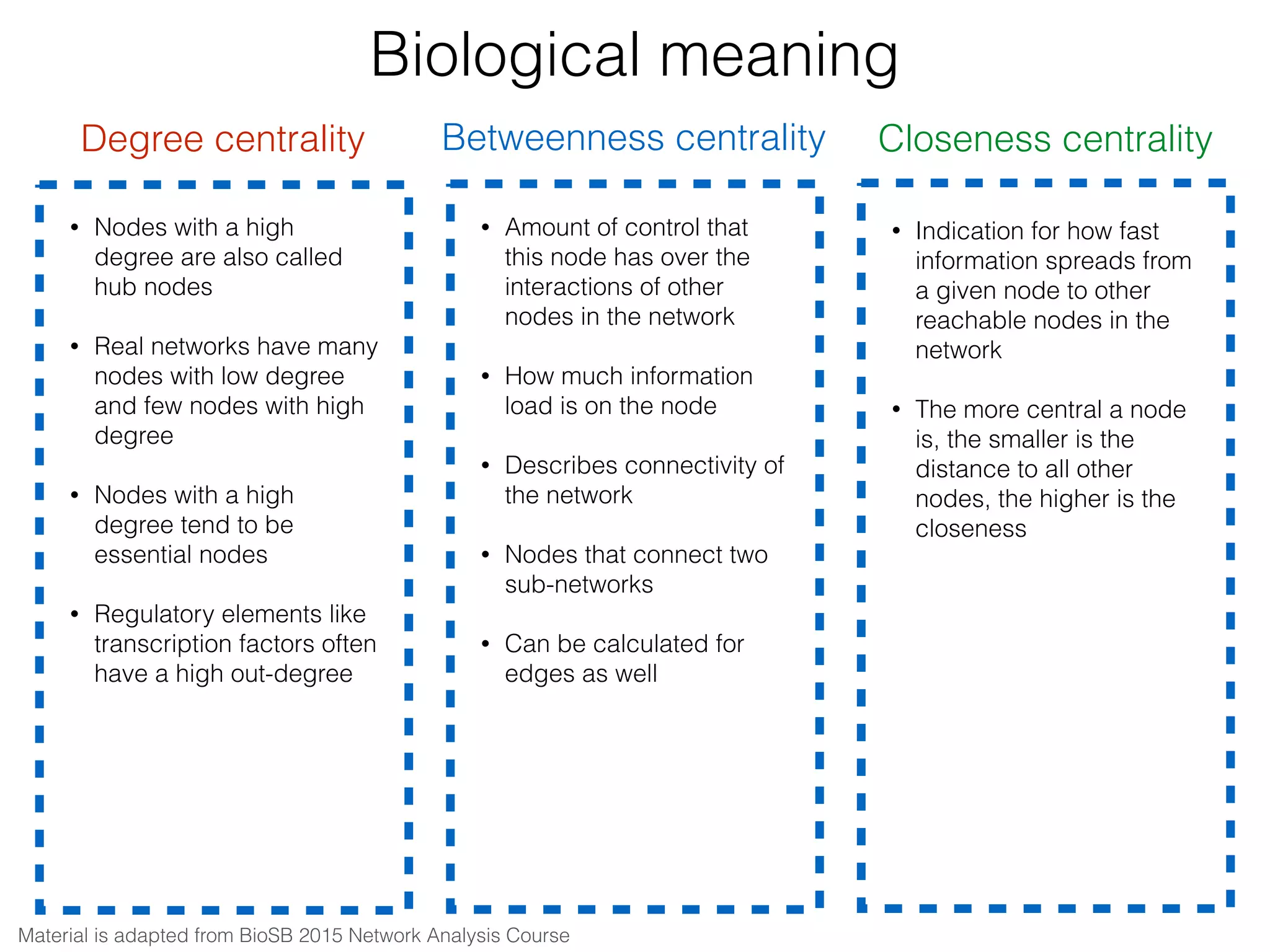
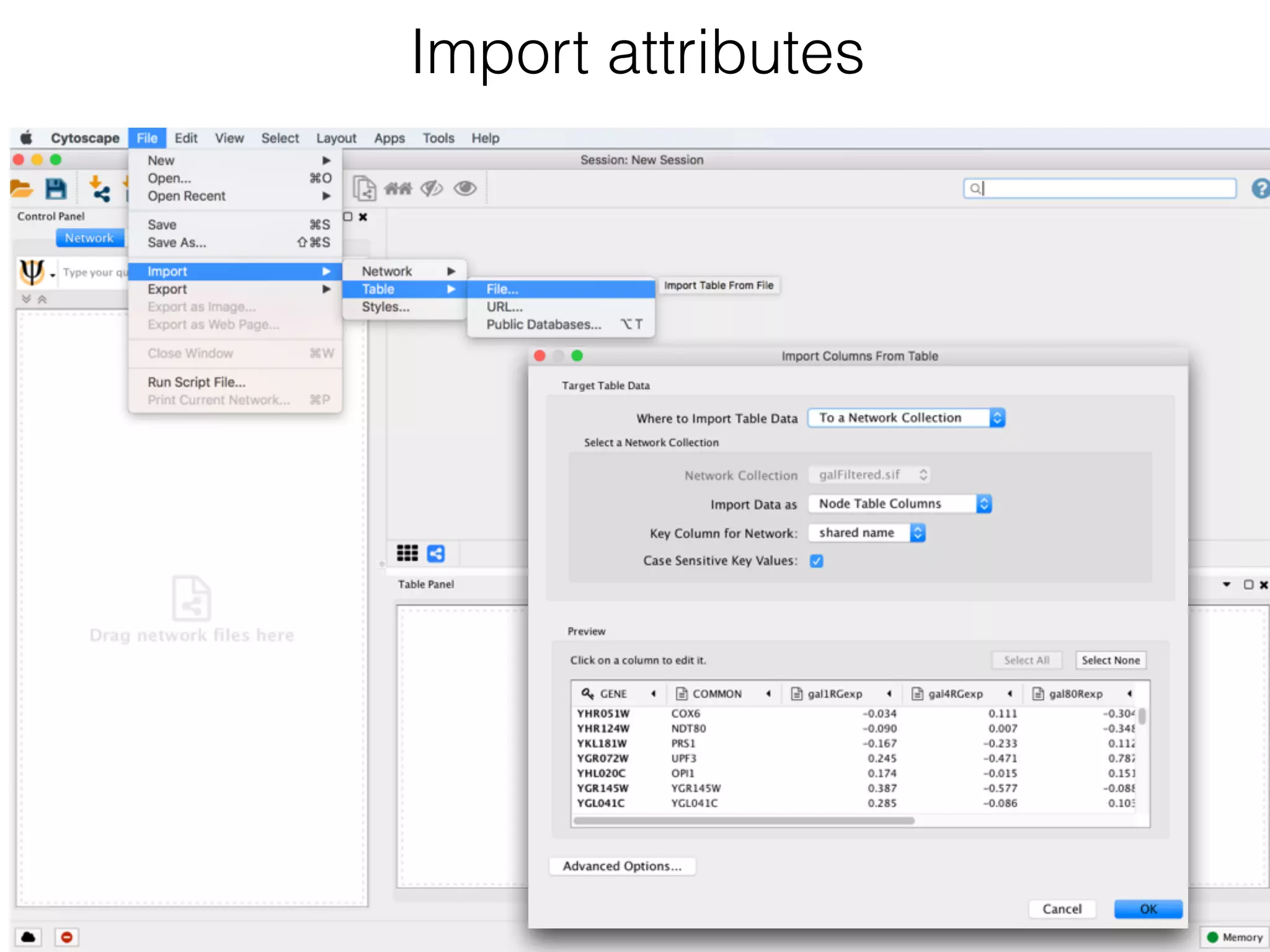
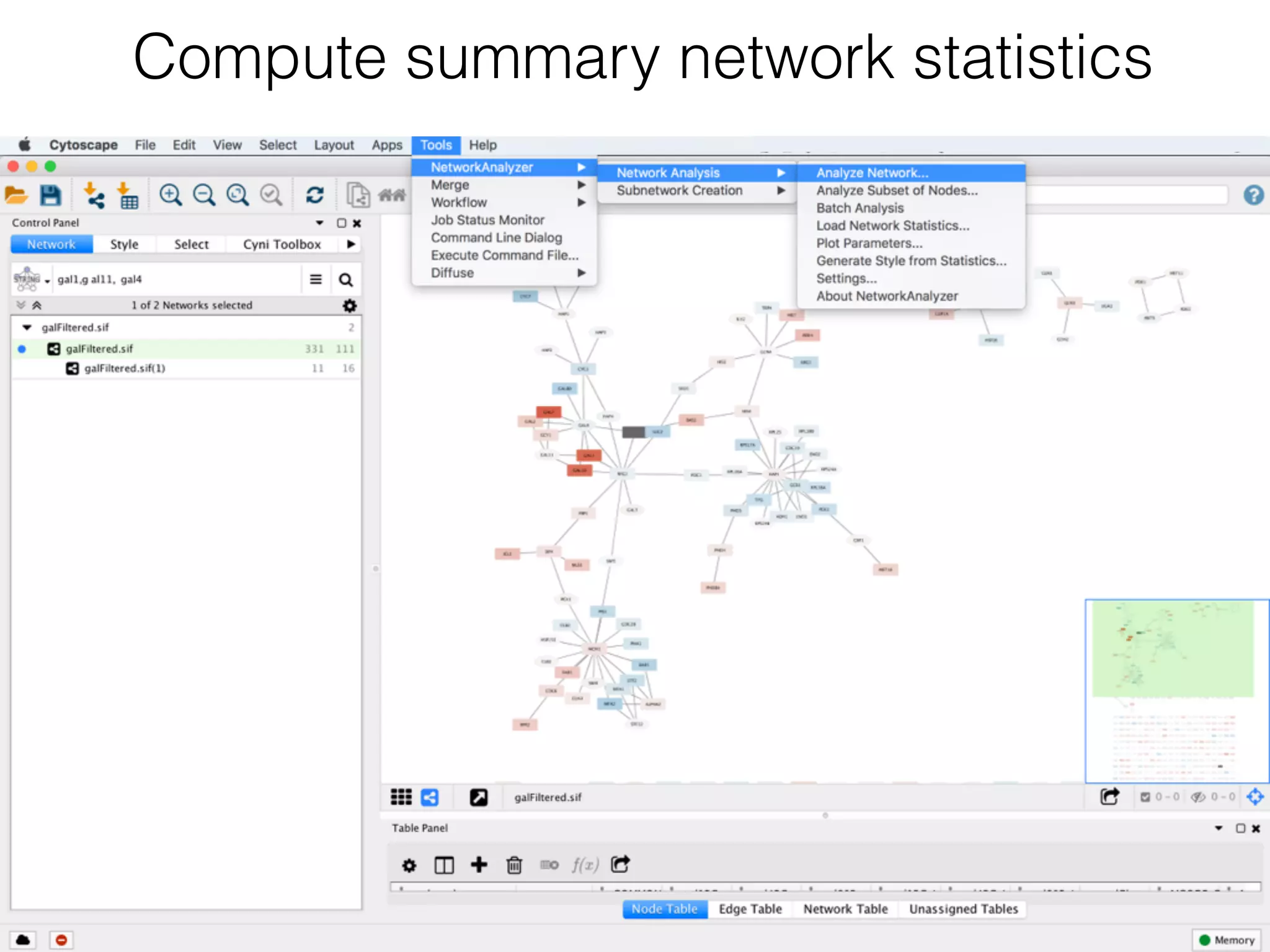
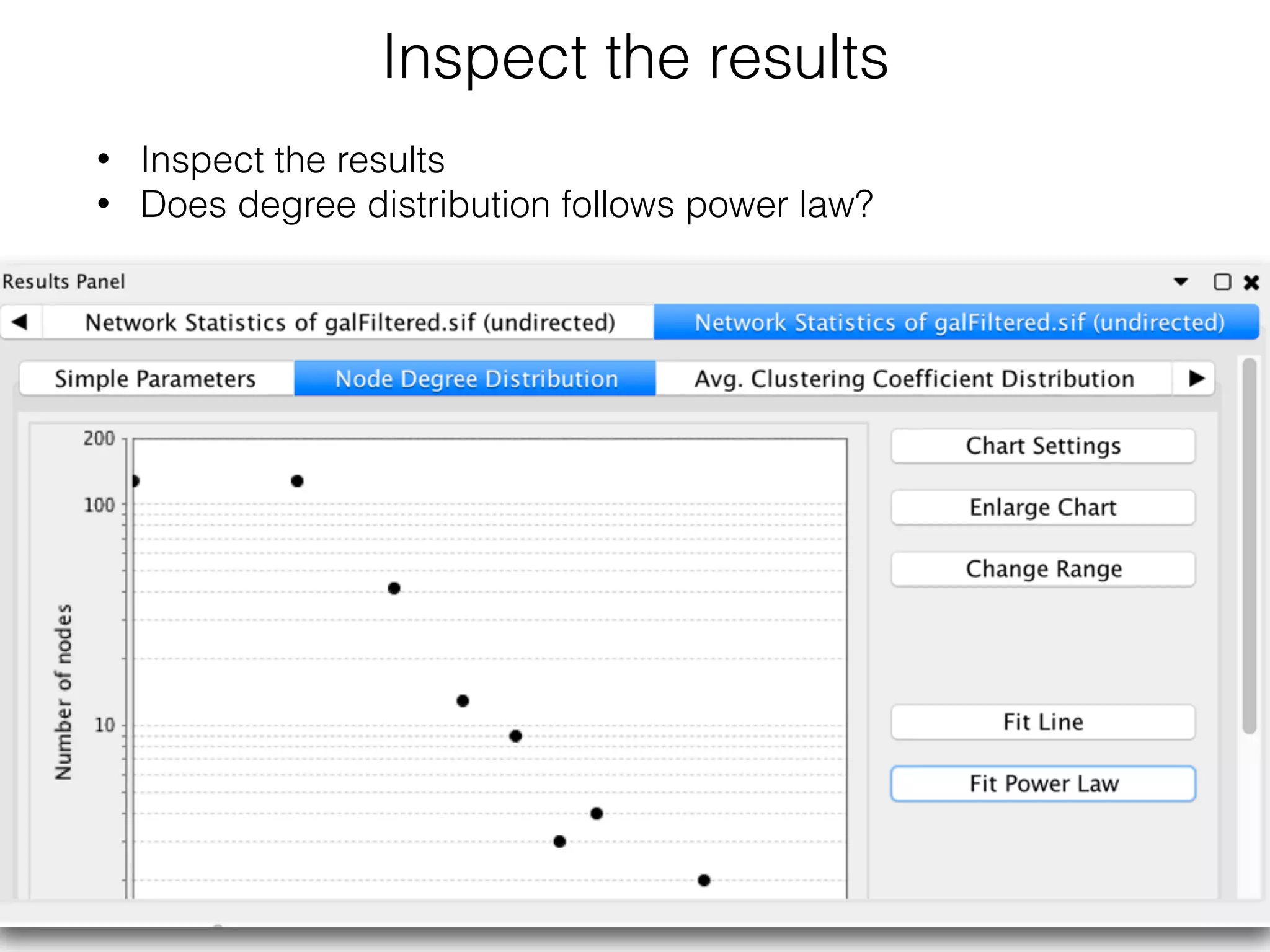
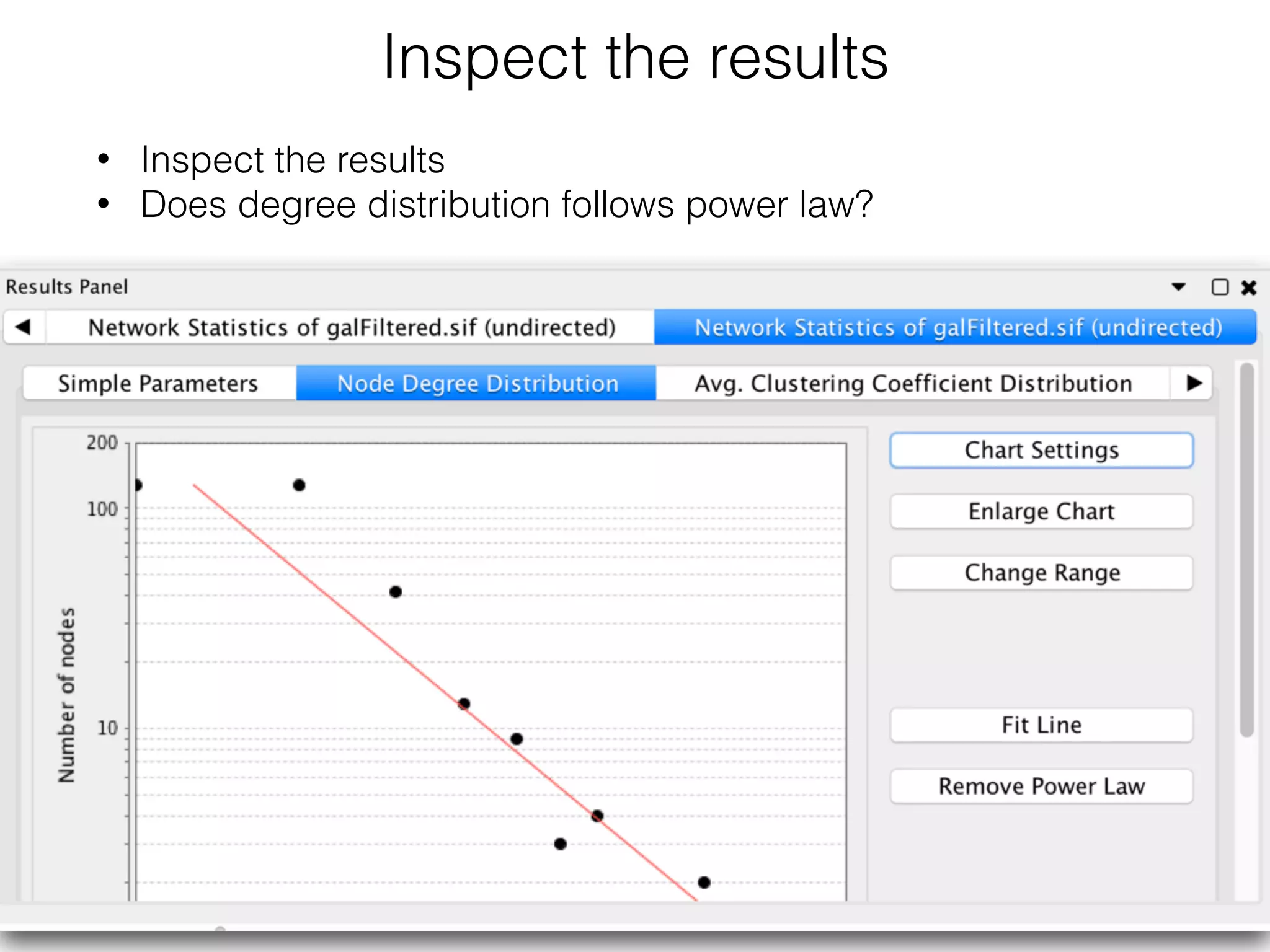
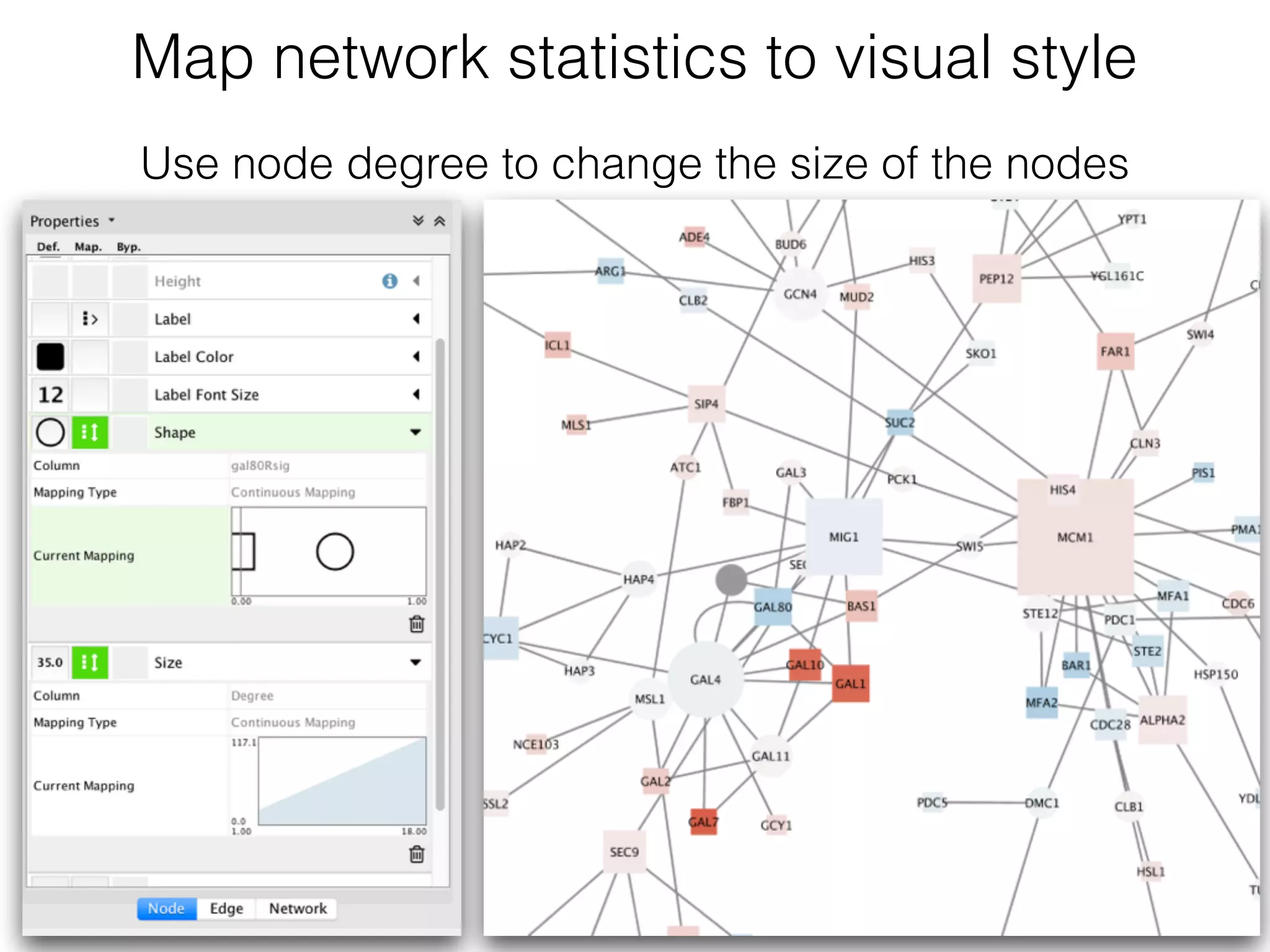
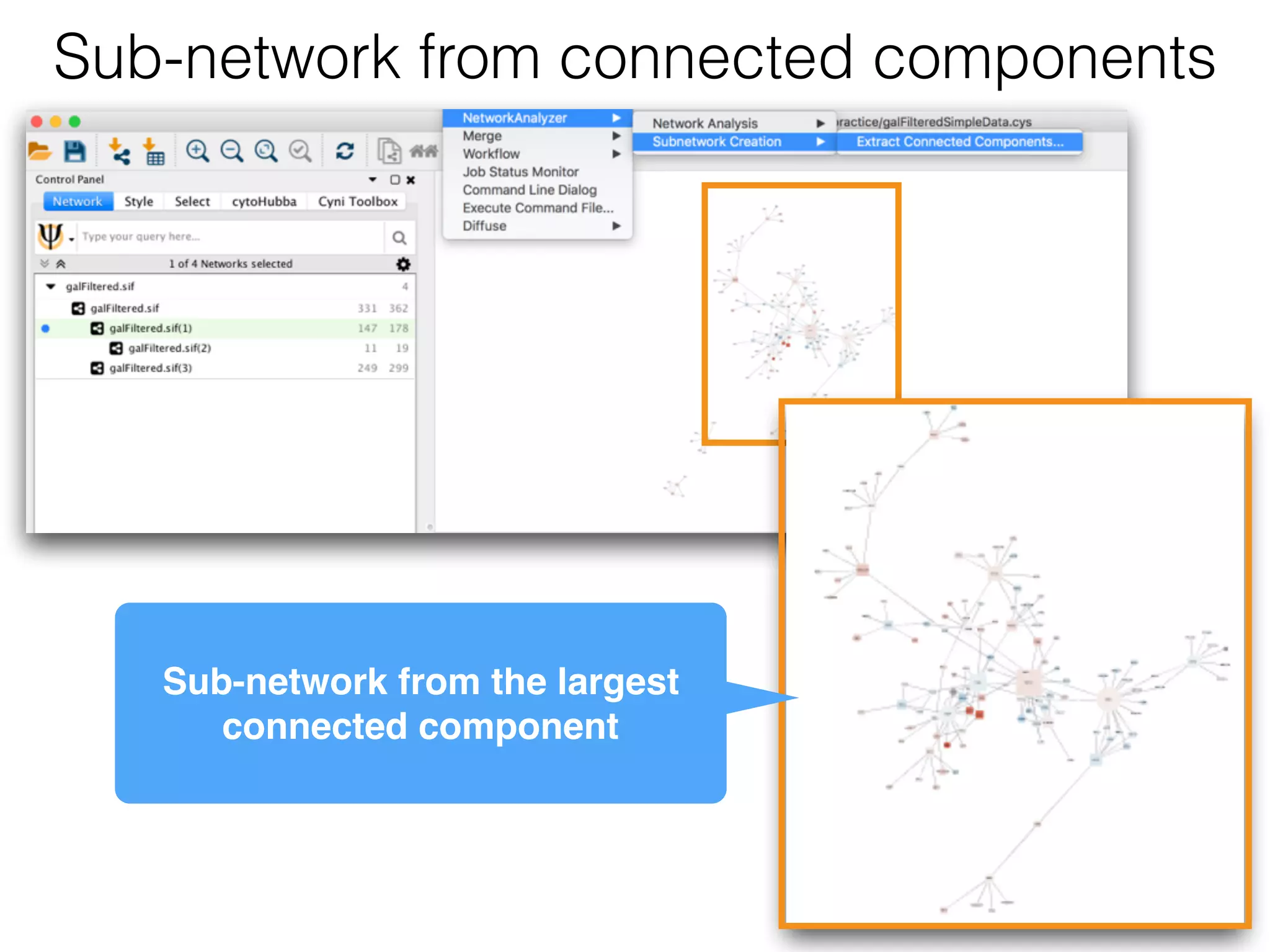
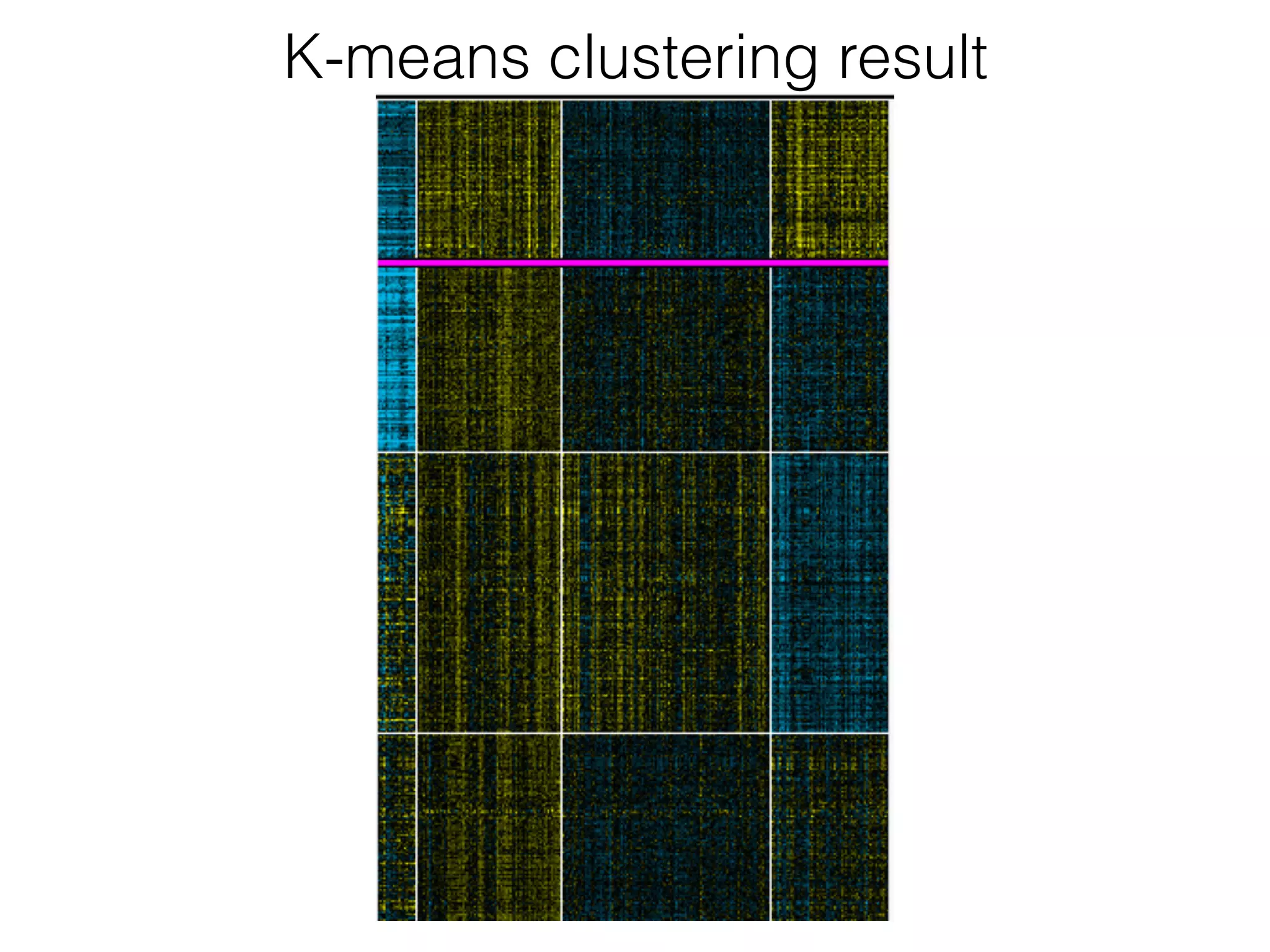
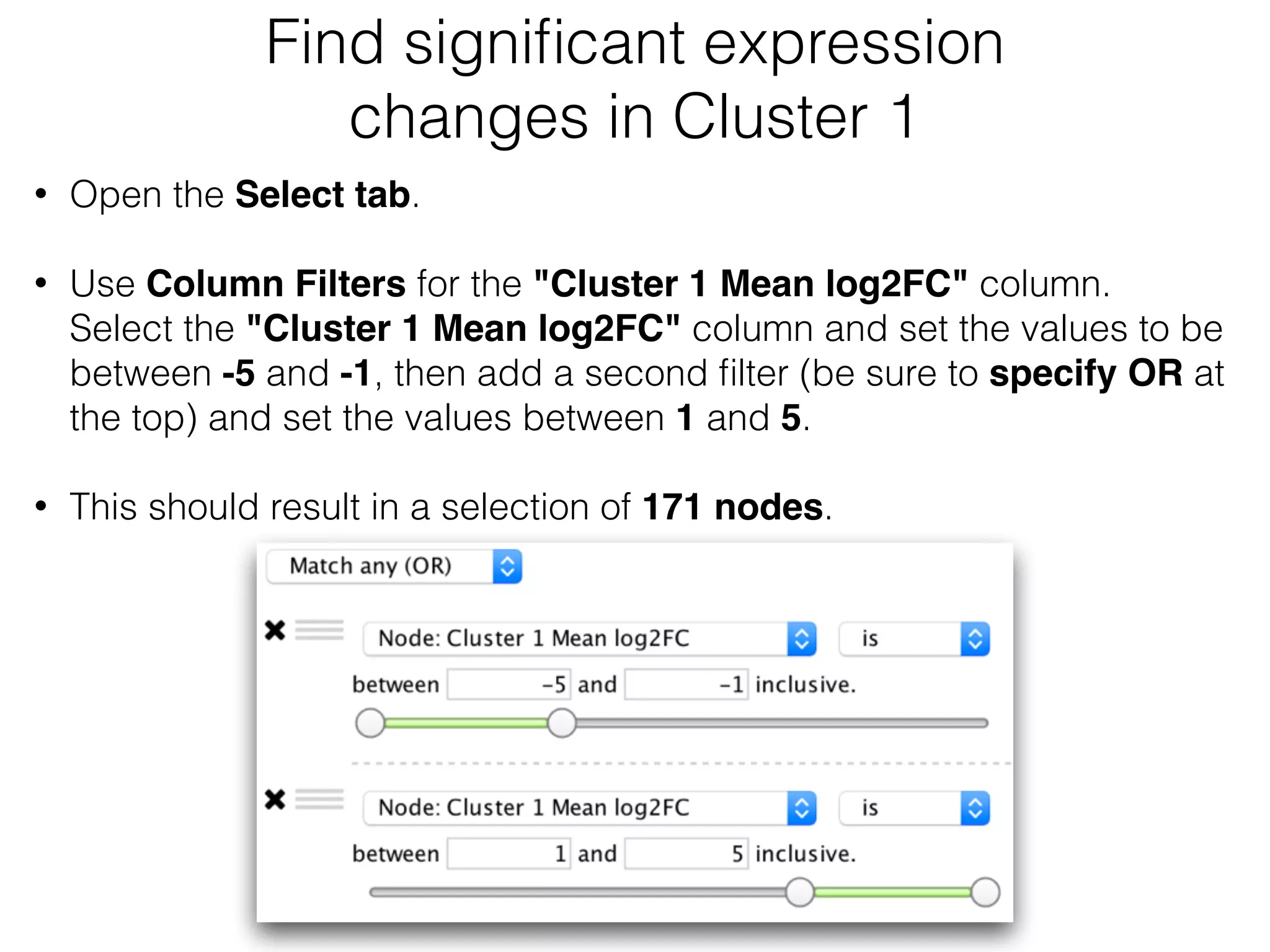
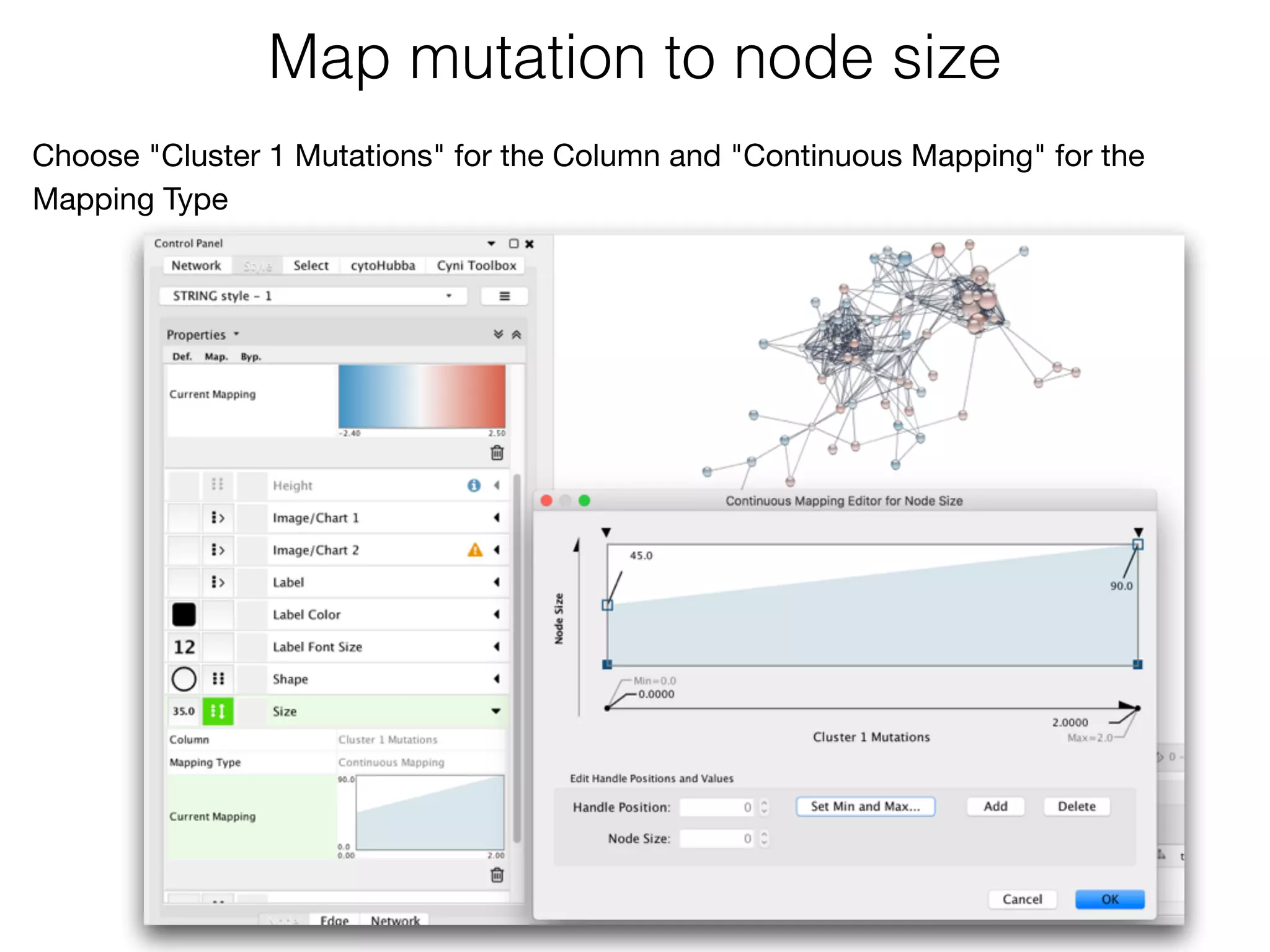
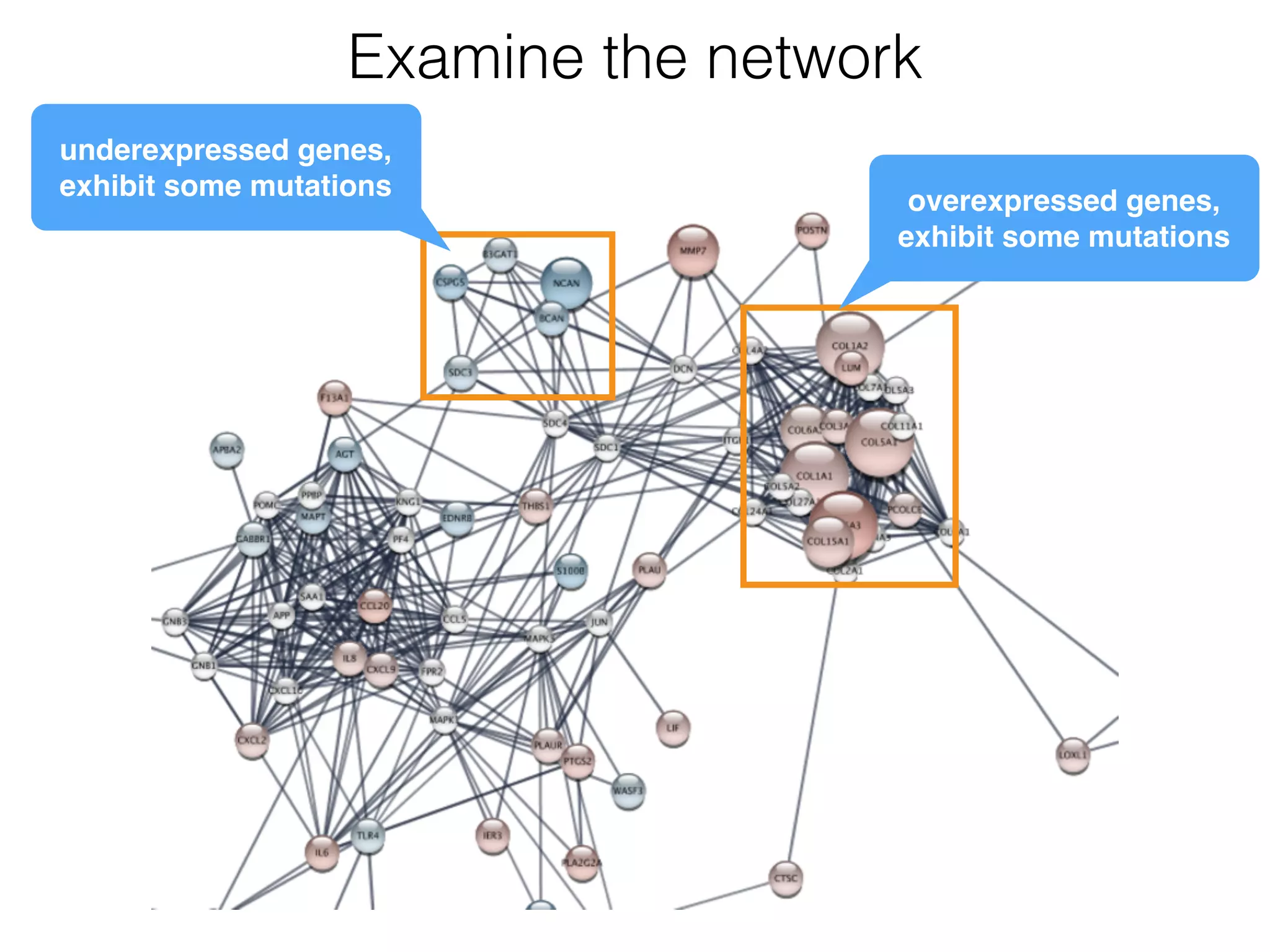
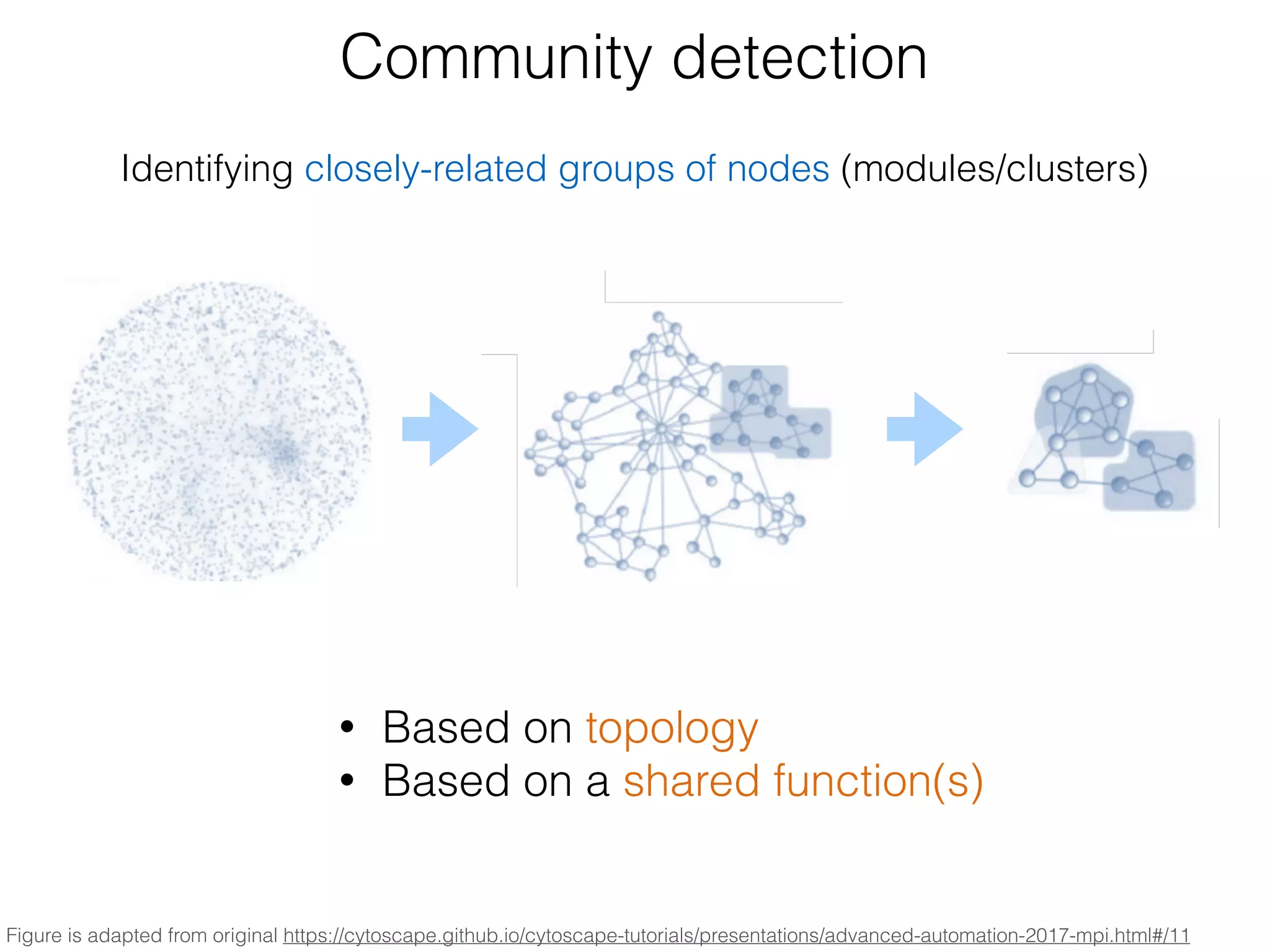
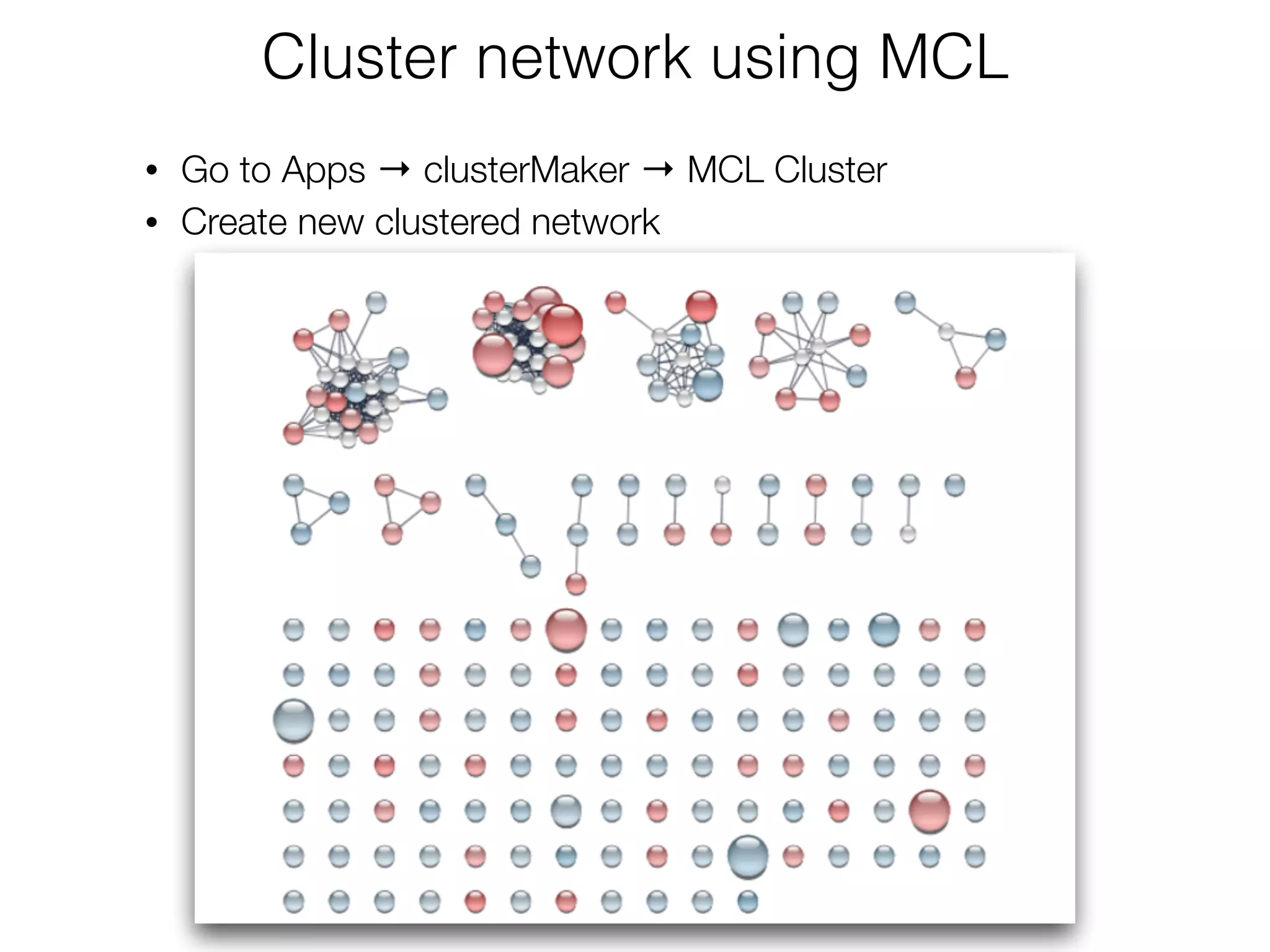
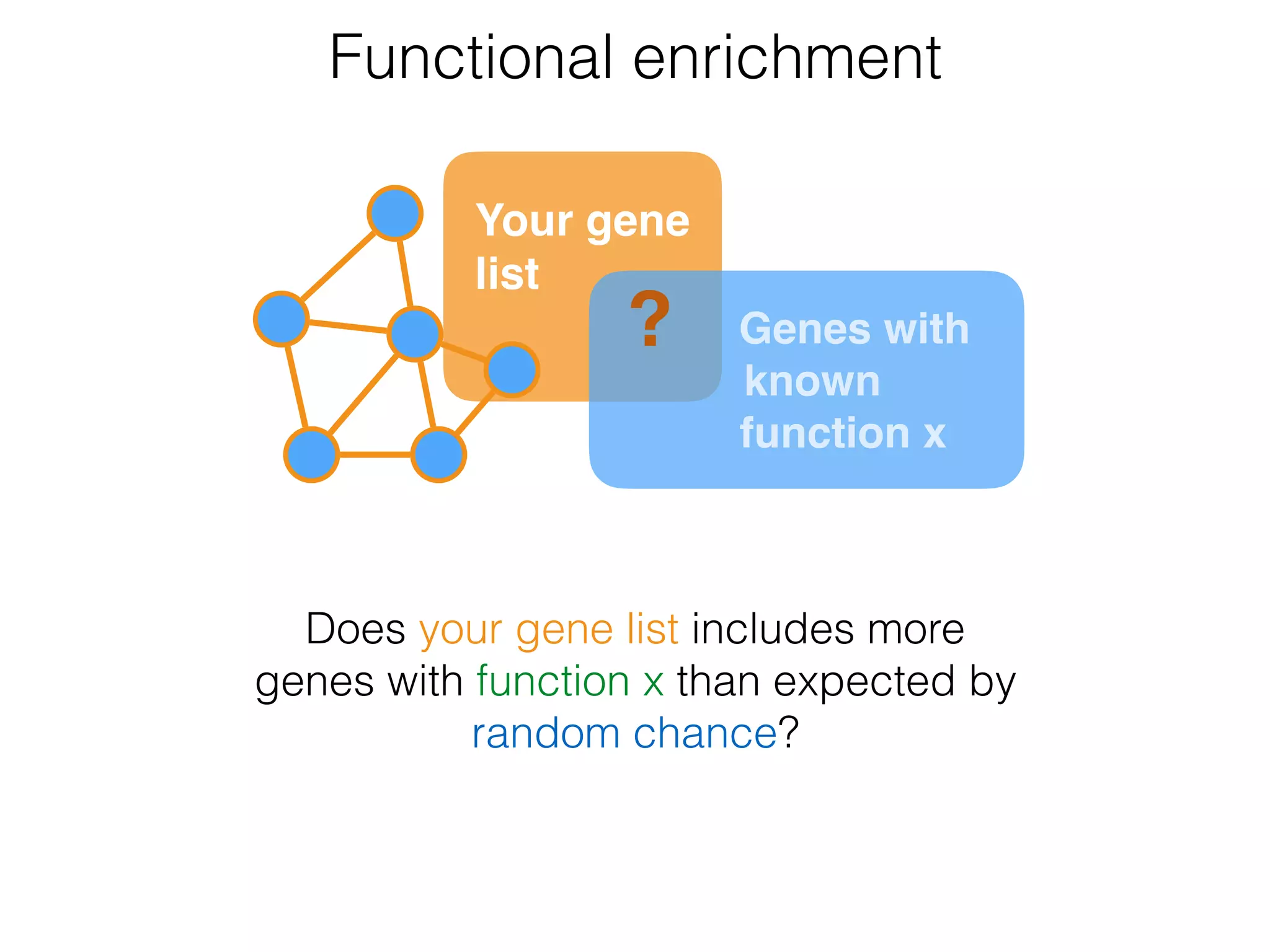
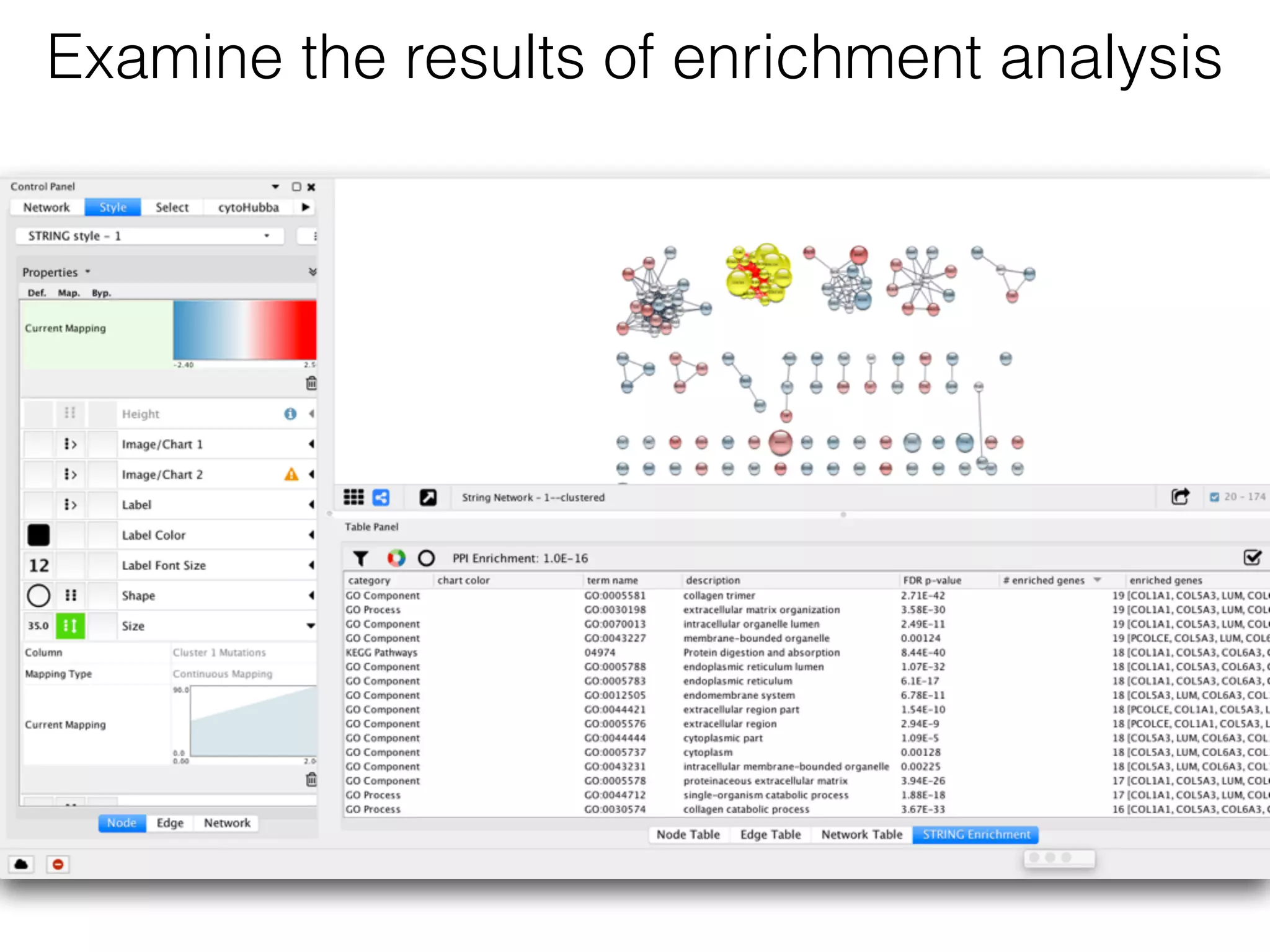
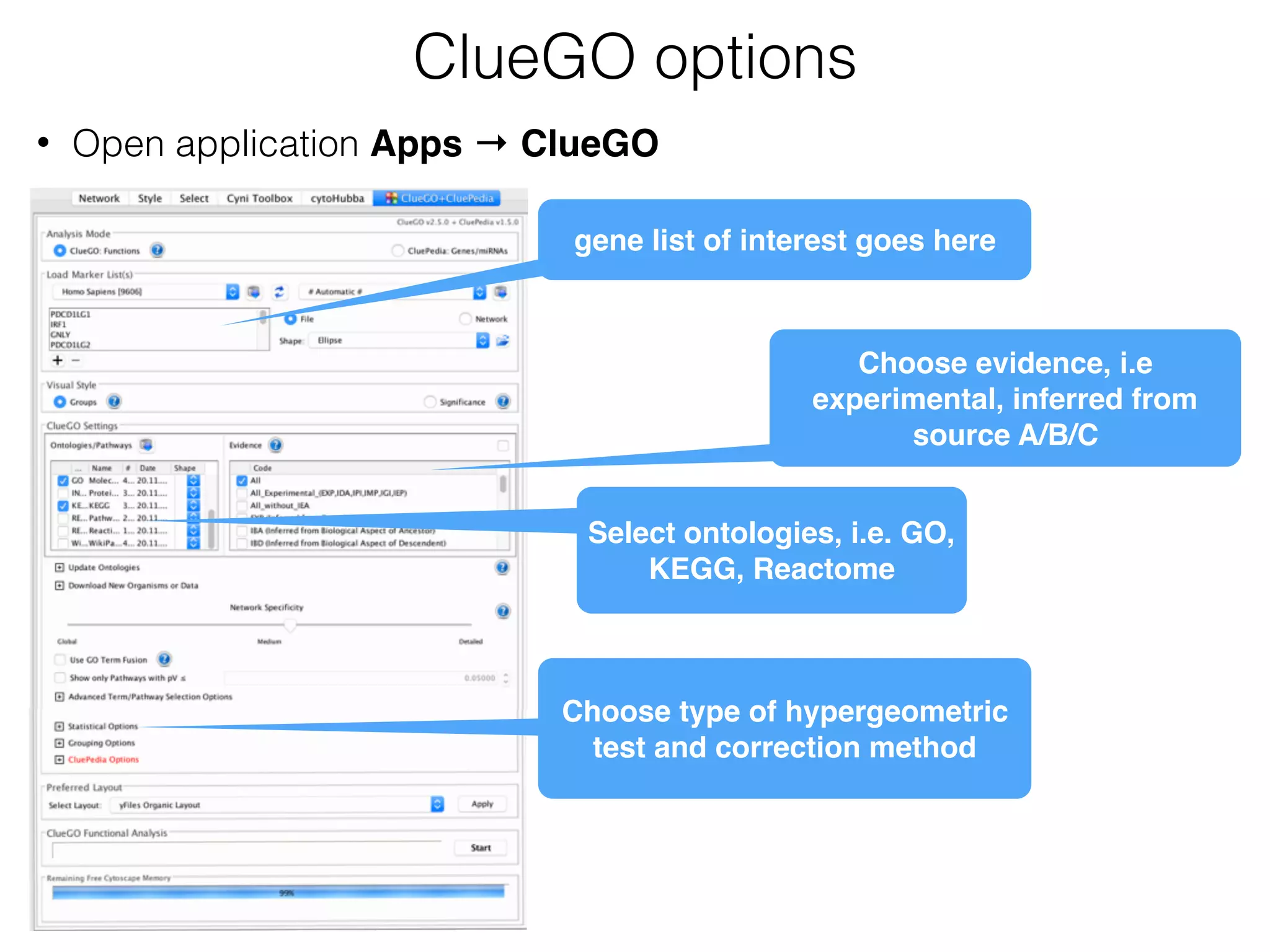
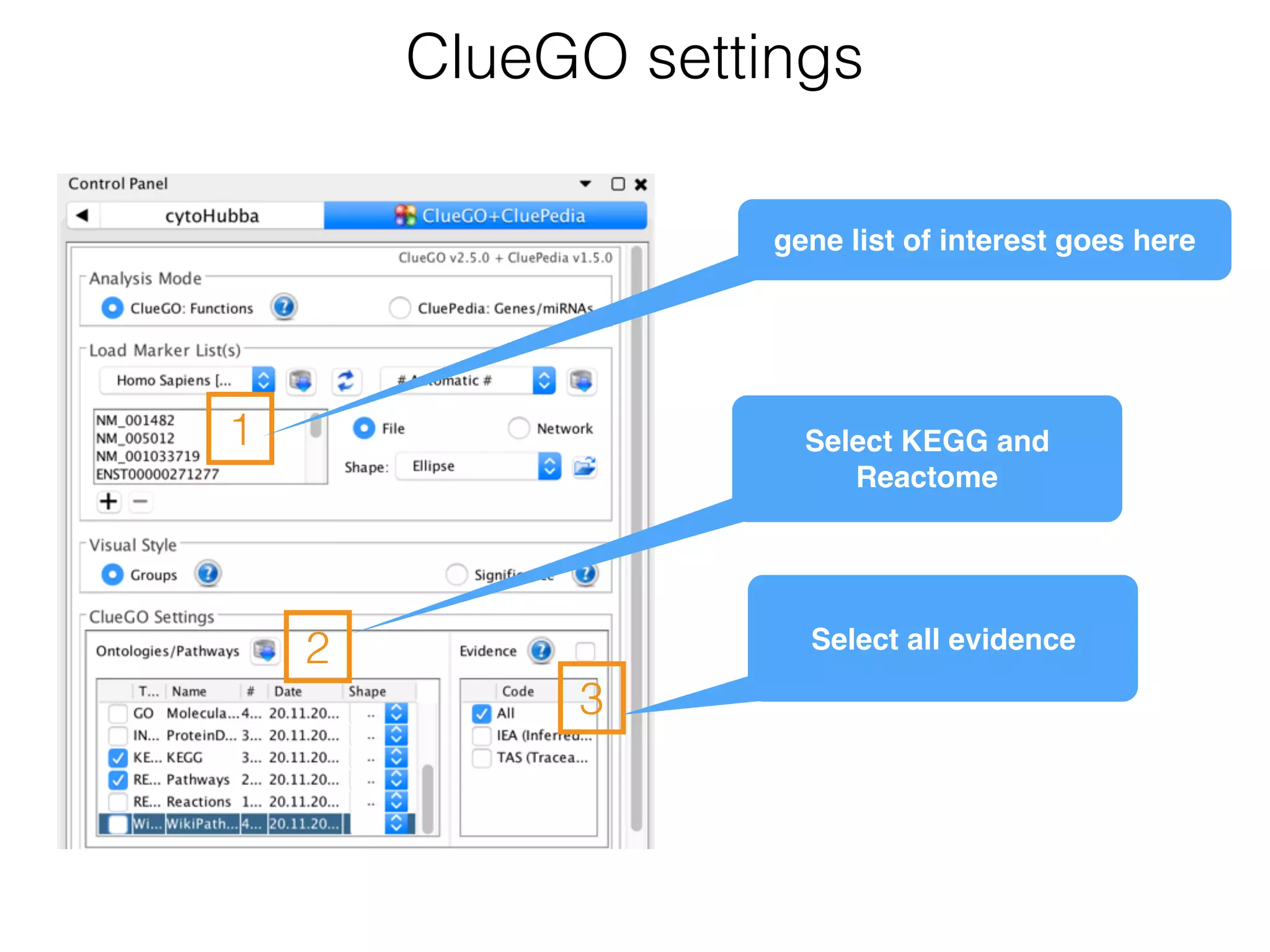
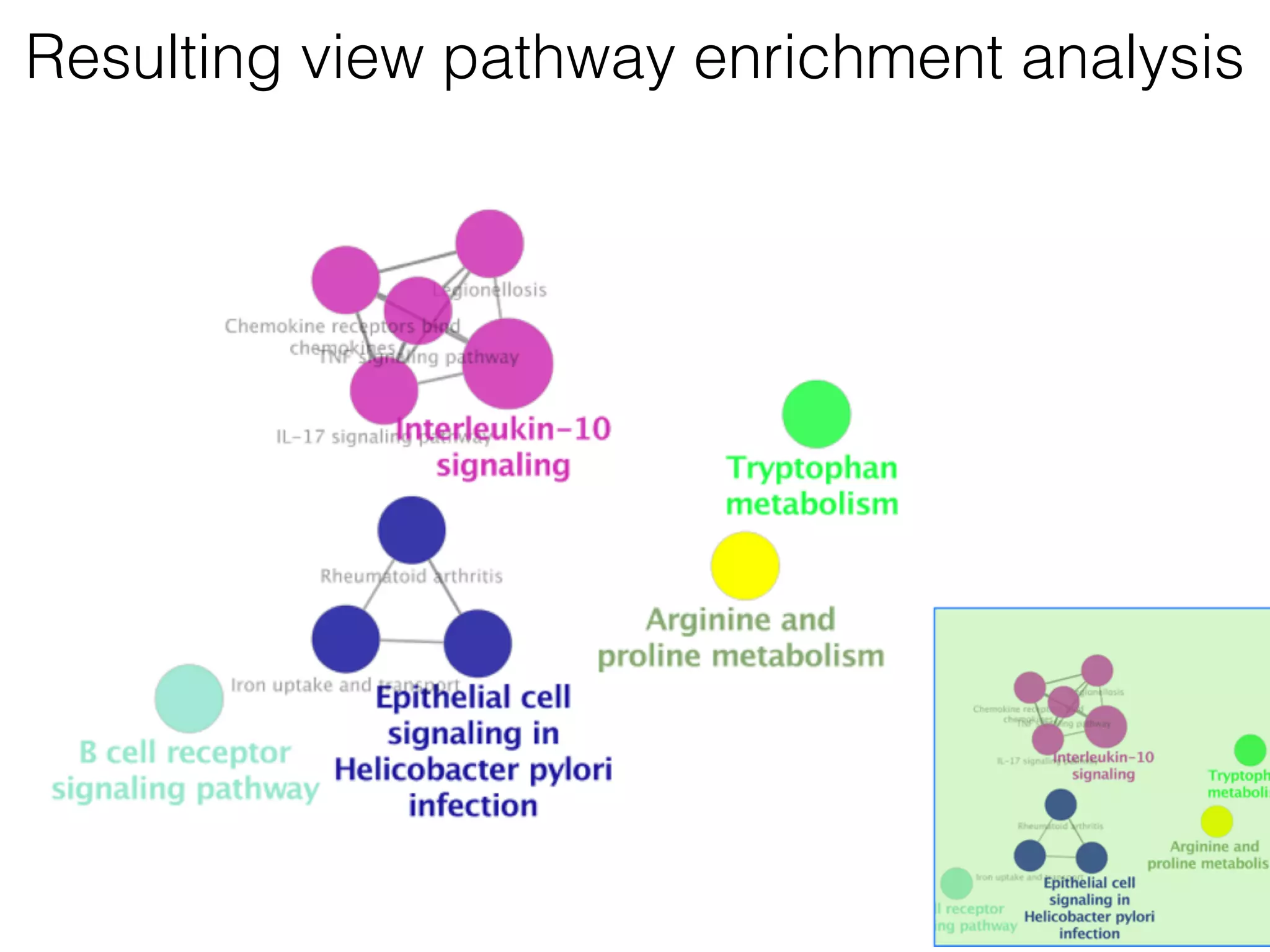
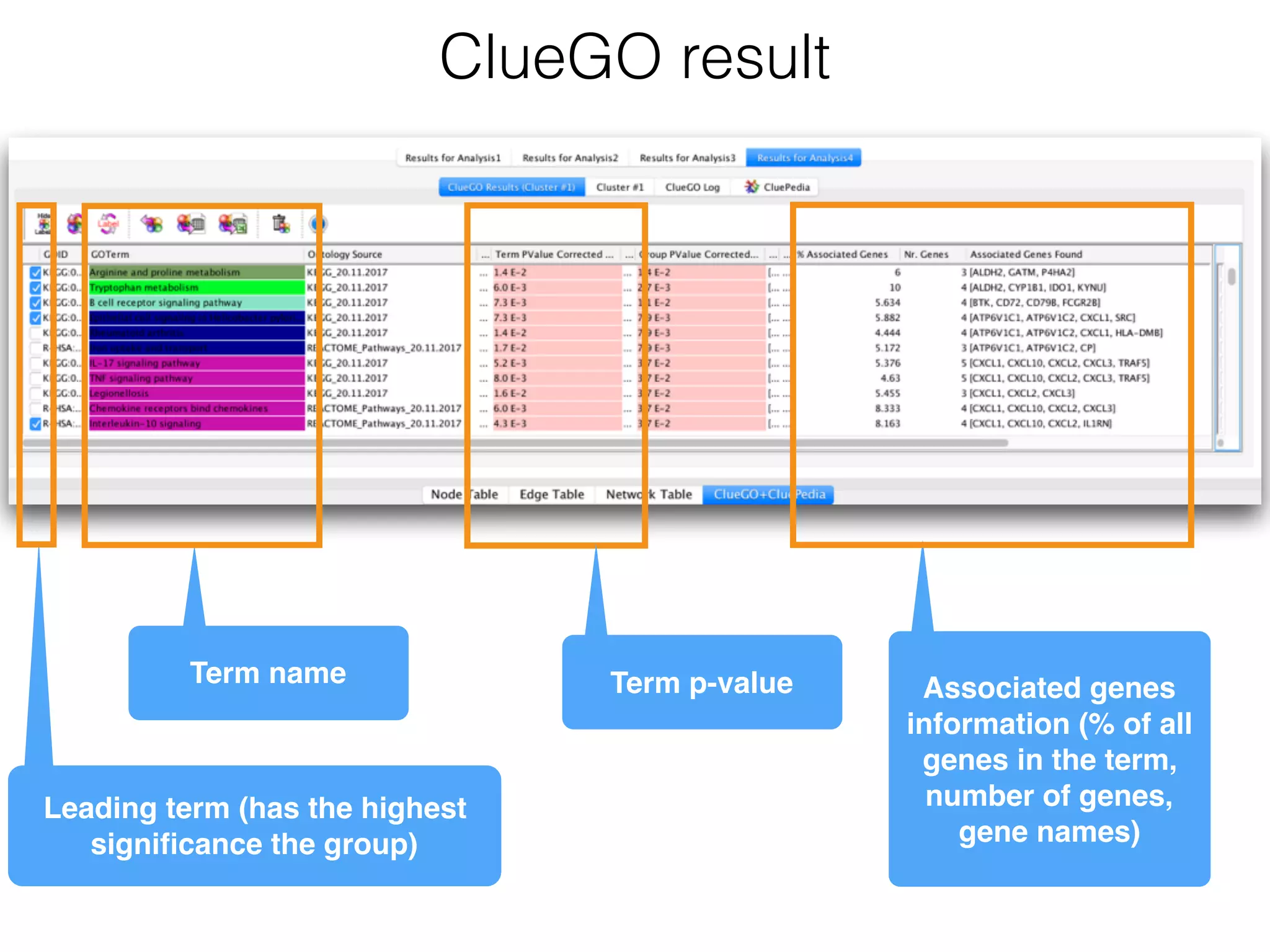
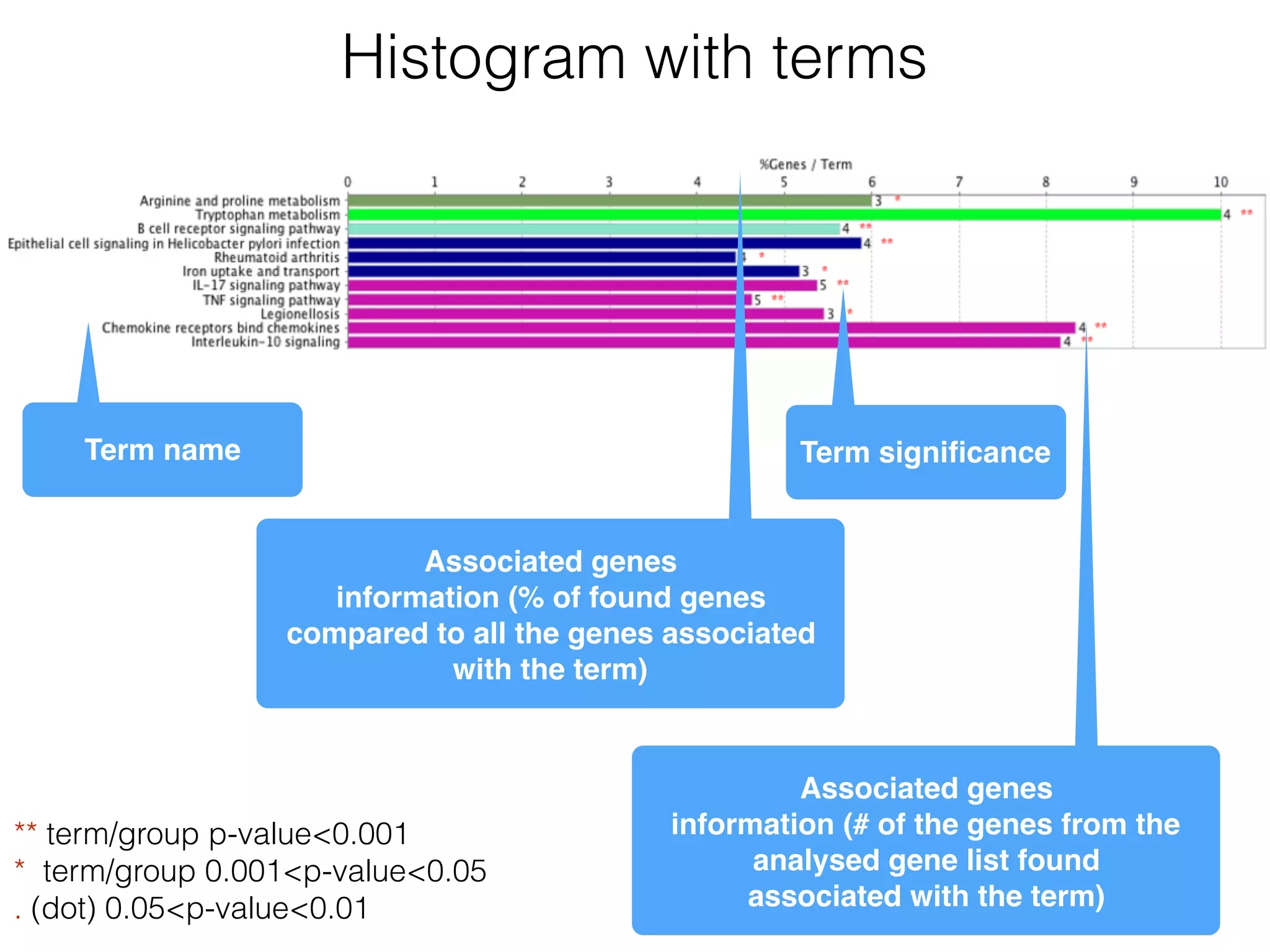
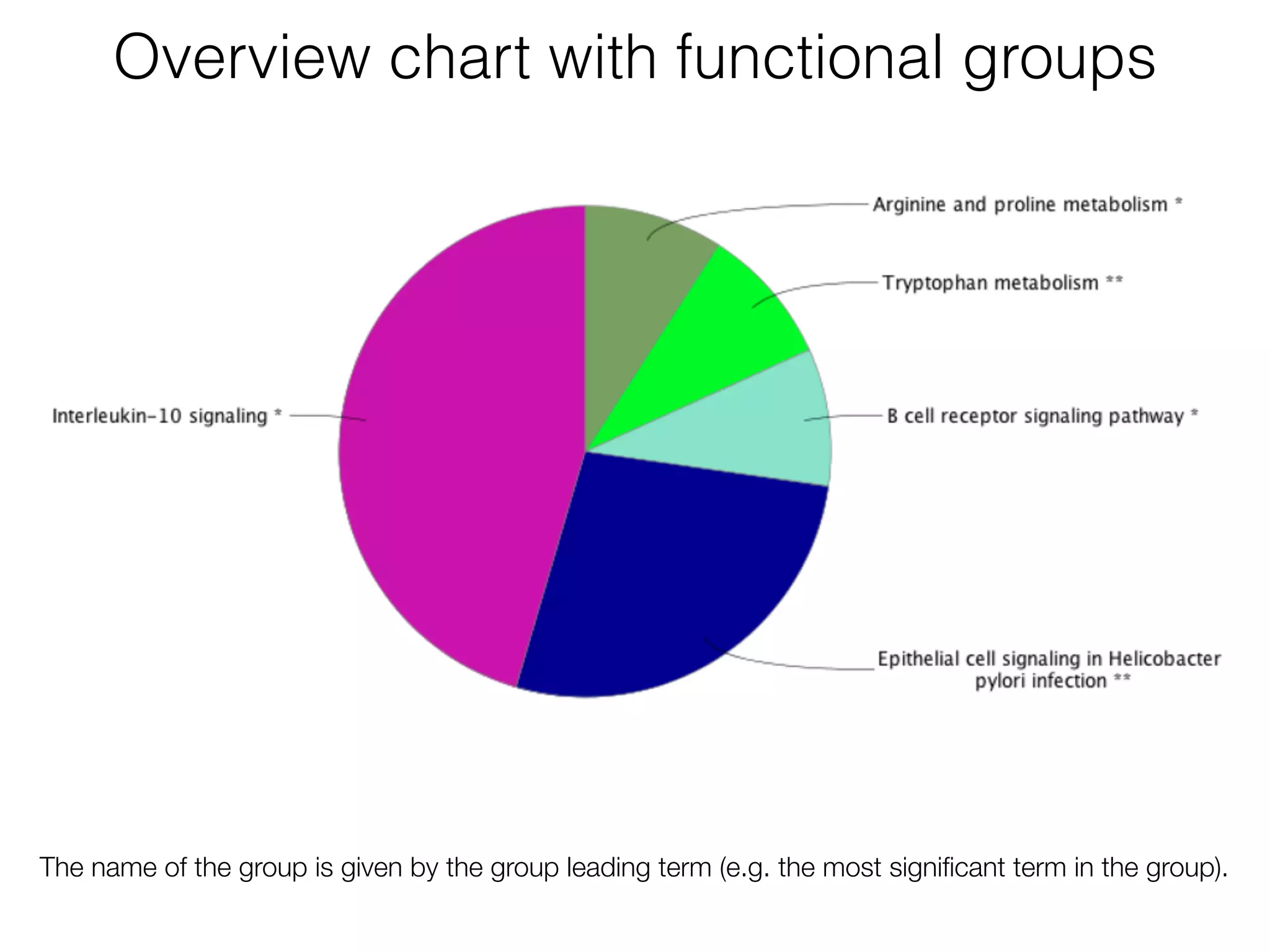
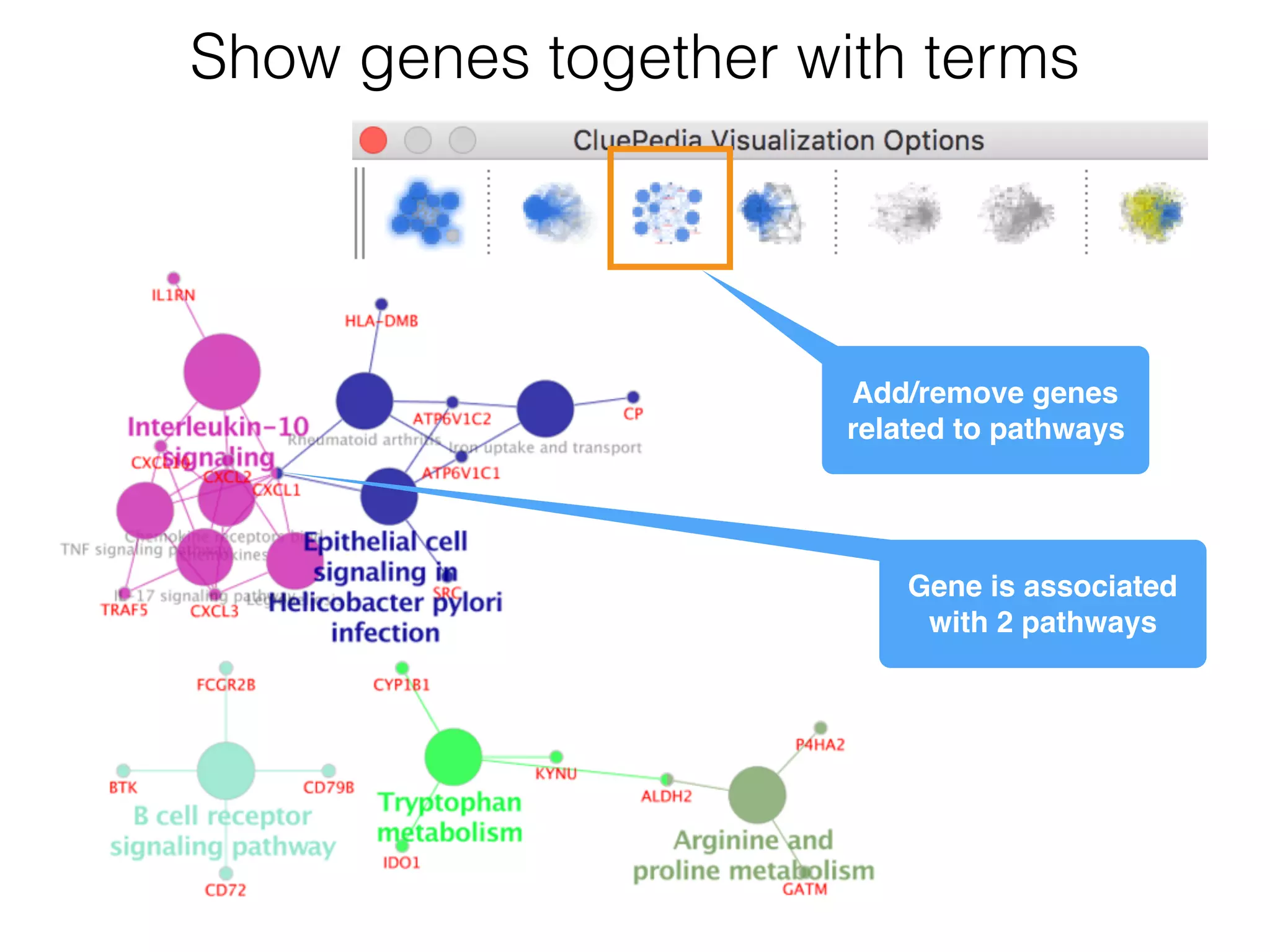
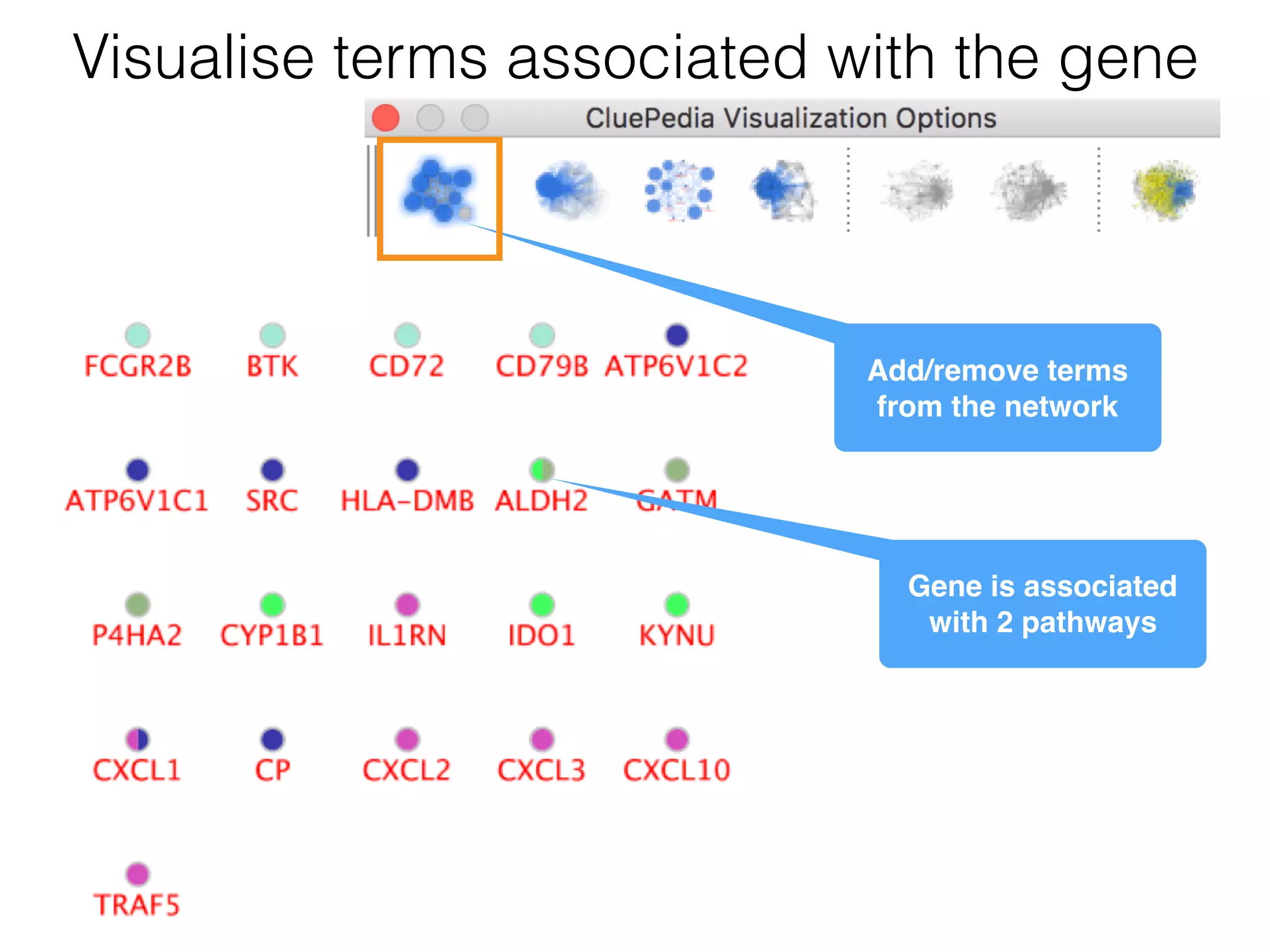
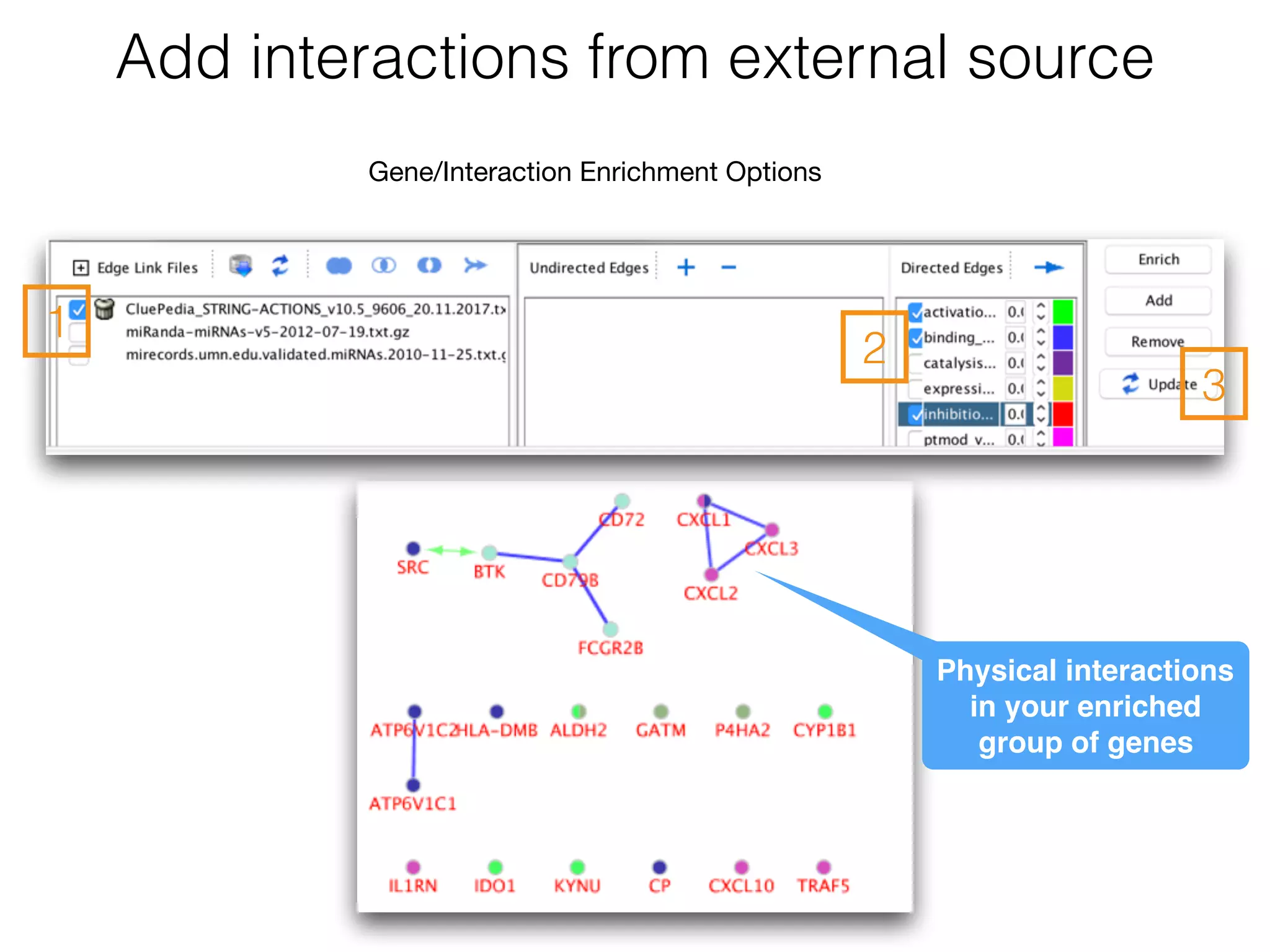
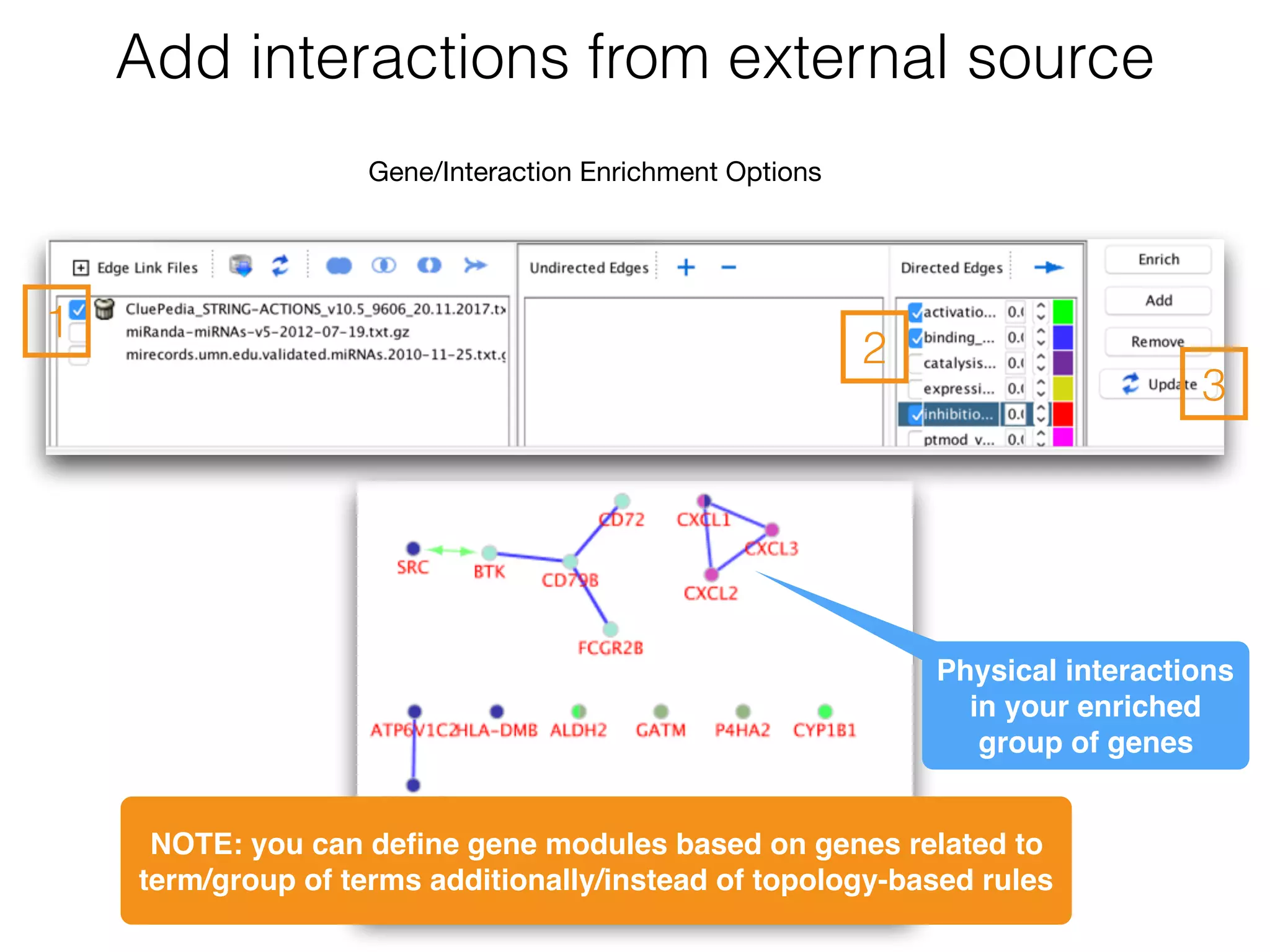
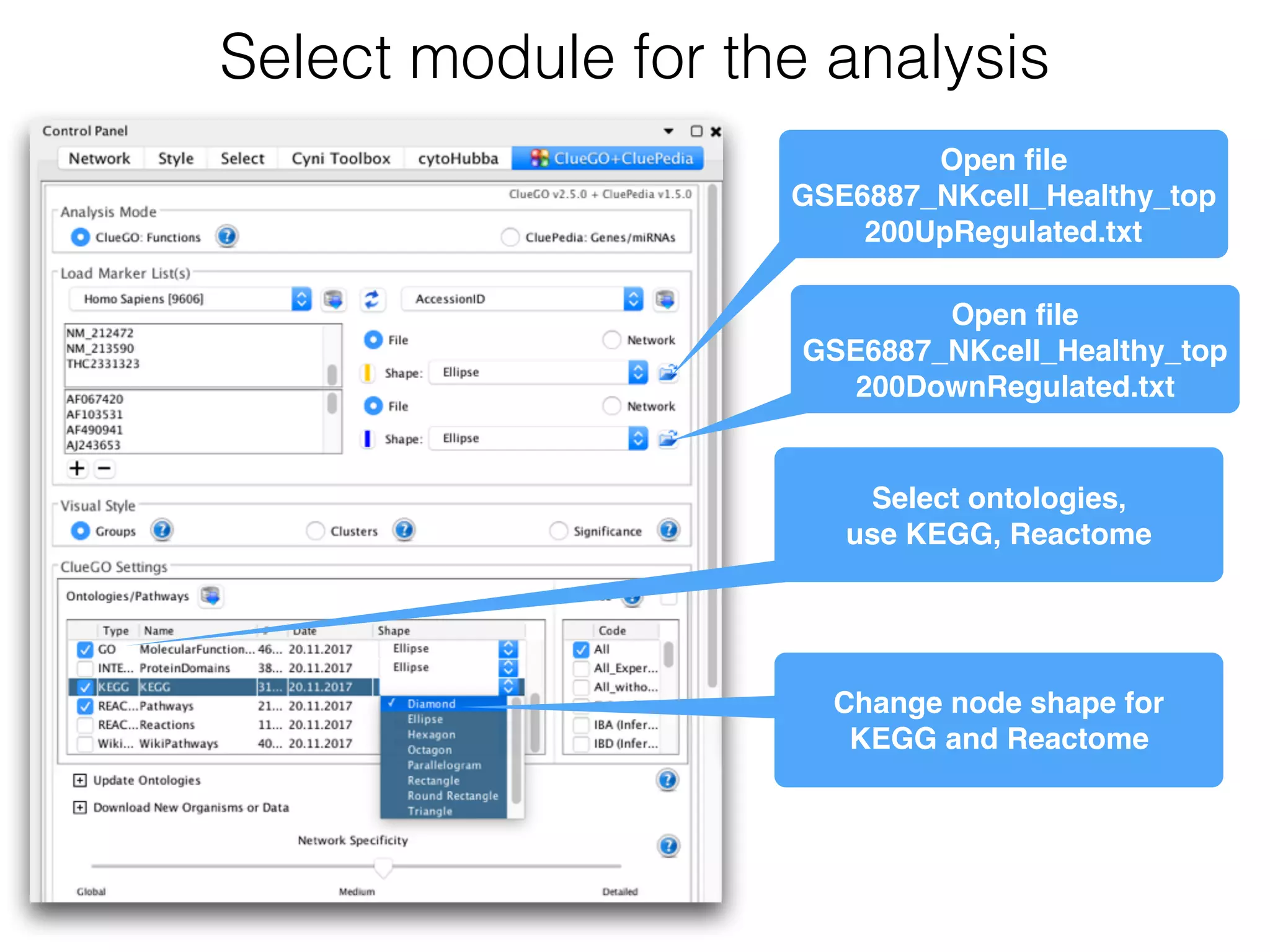
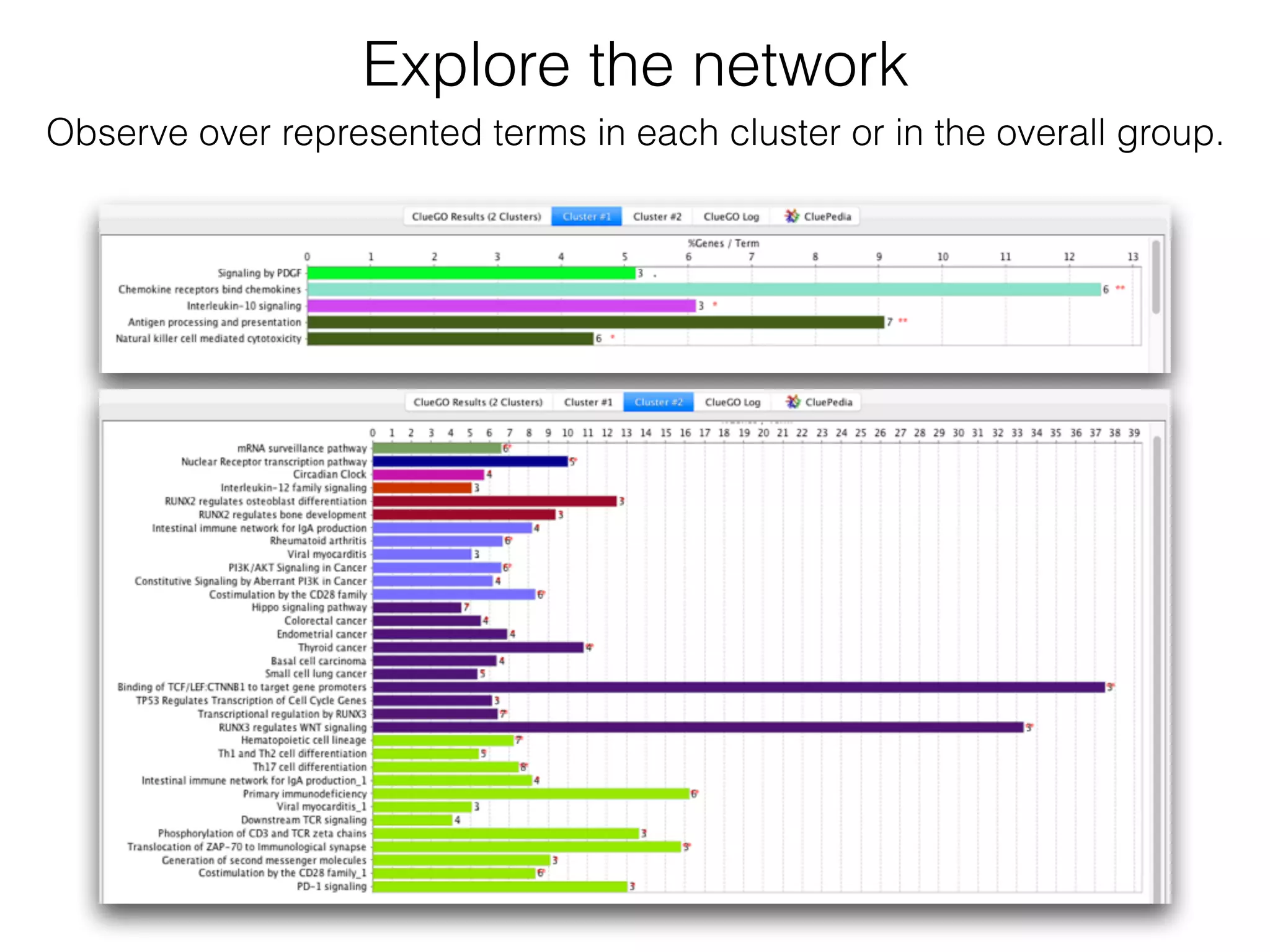
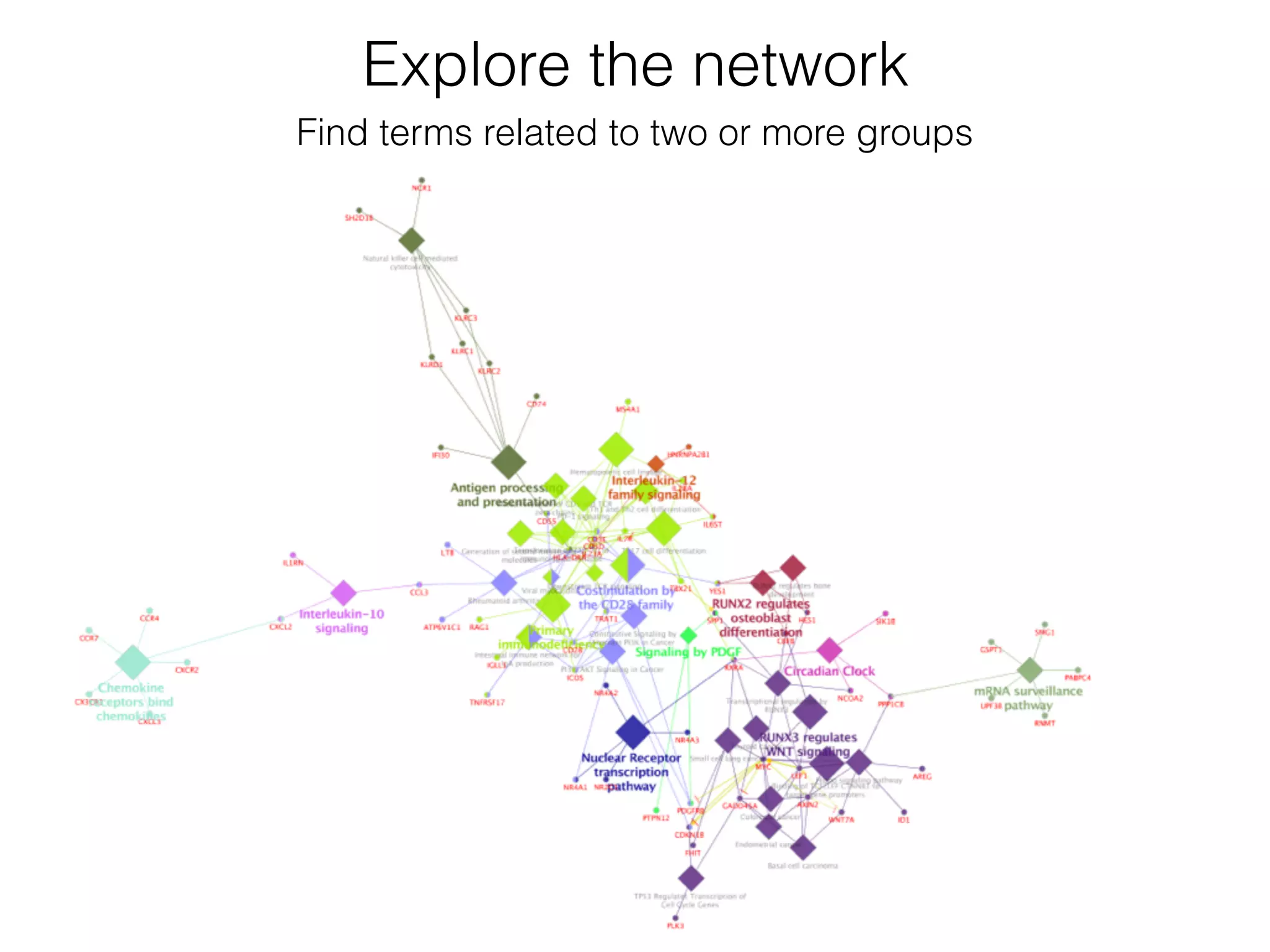
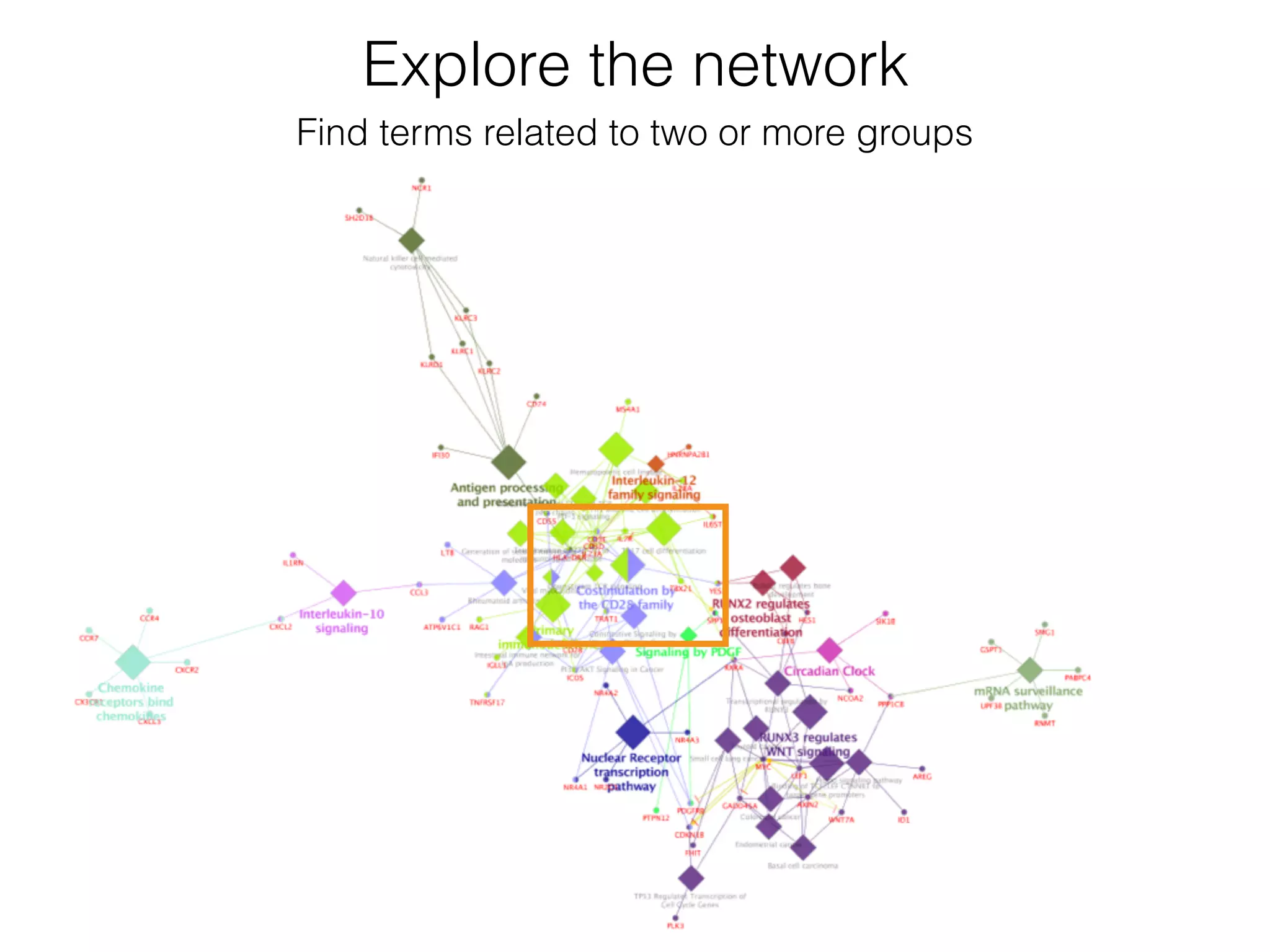
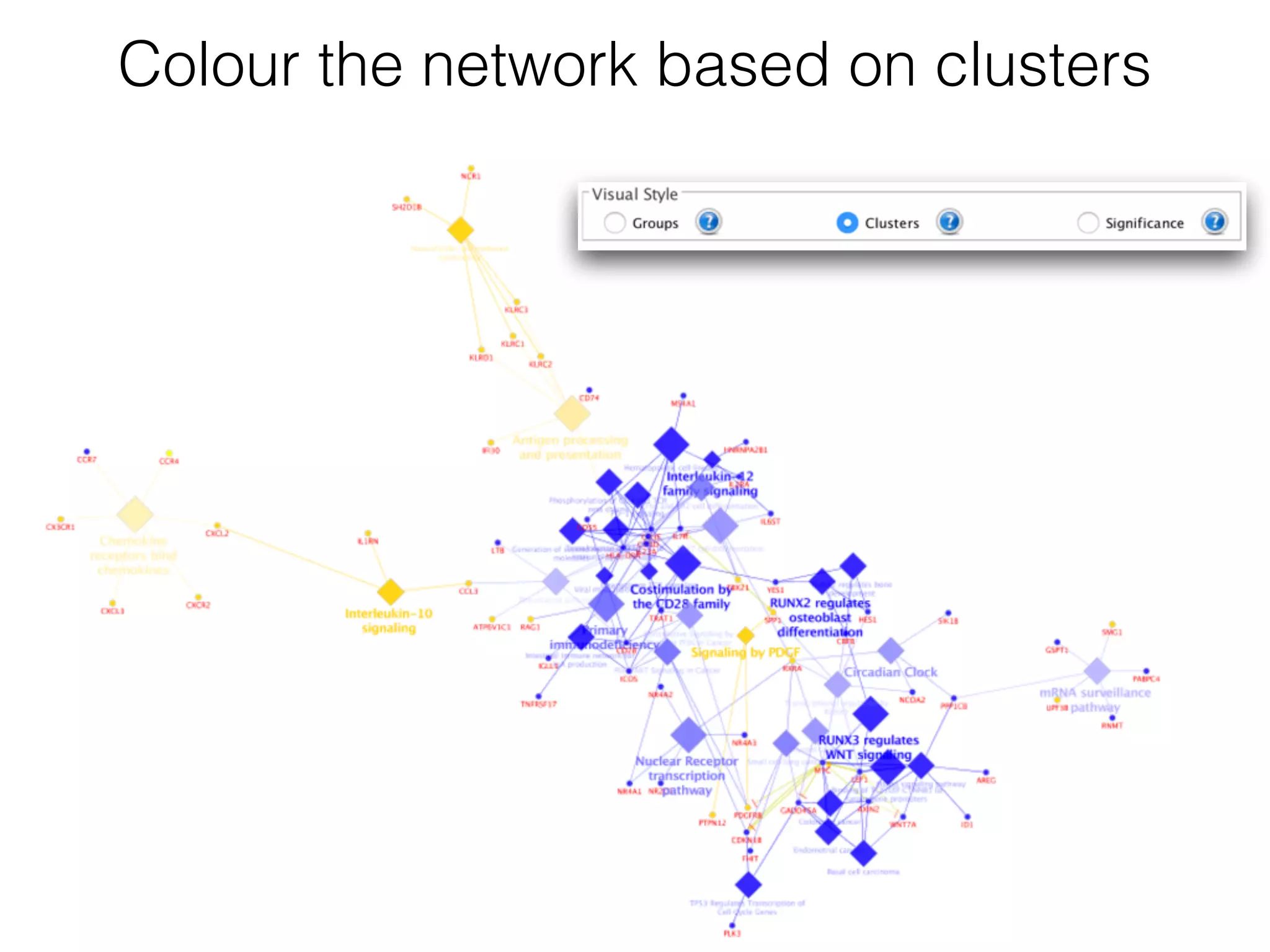
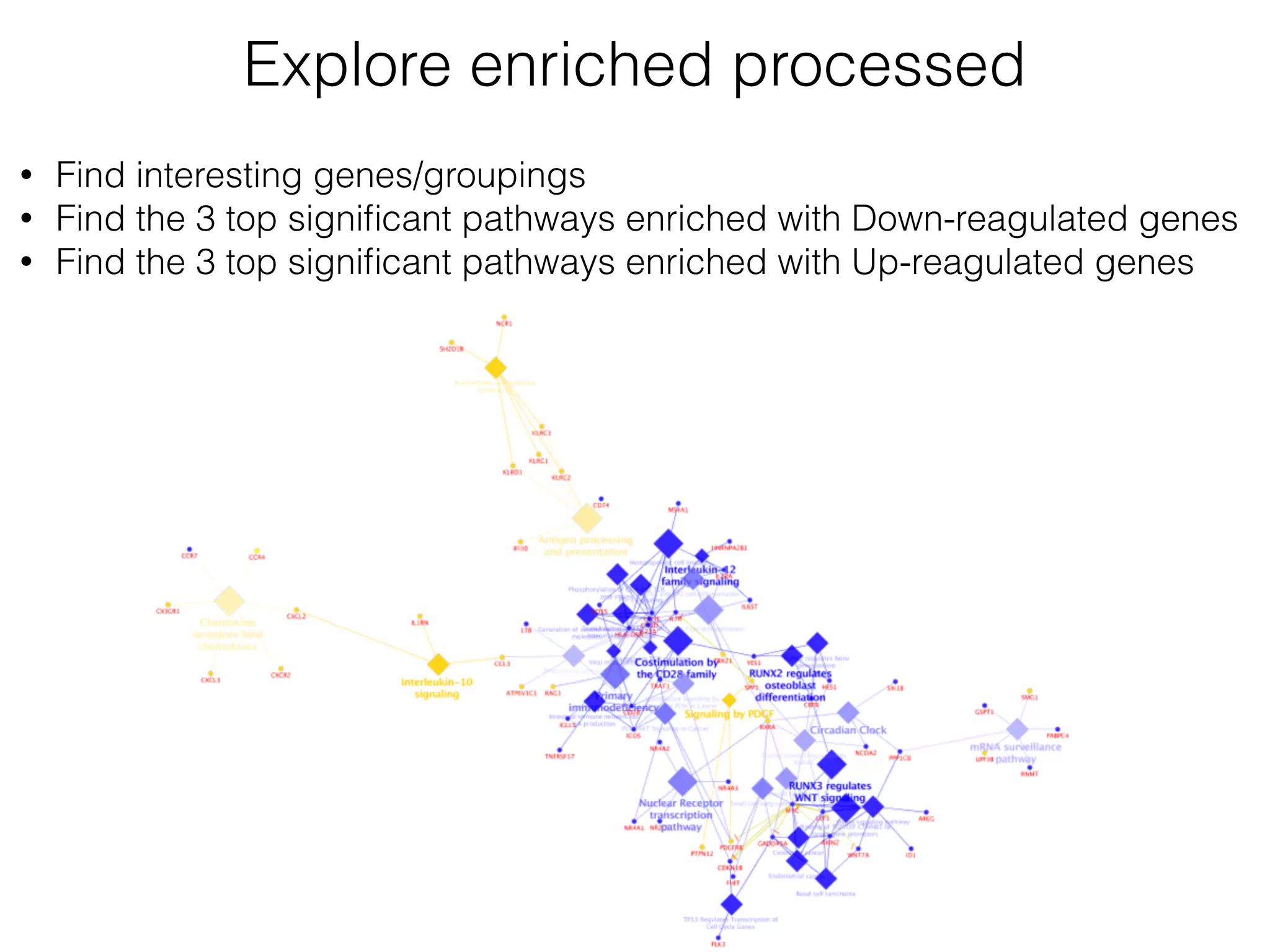
The document outlines a practice session on using Cytoscape for biological data visualization and analysis, covering core concepts such as networks, nodes, and edges. It details prerequisites, software installation, data import, and visualization techniques, as well as various analysis methods, including statistical computations and functional enrichment analysis. The session emphasizes practical exercises for understanding network data and interpreting biological relationships through visual tools.