Downloaded 296 times










































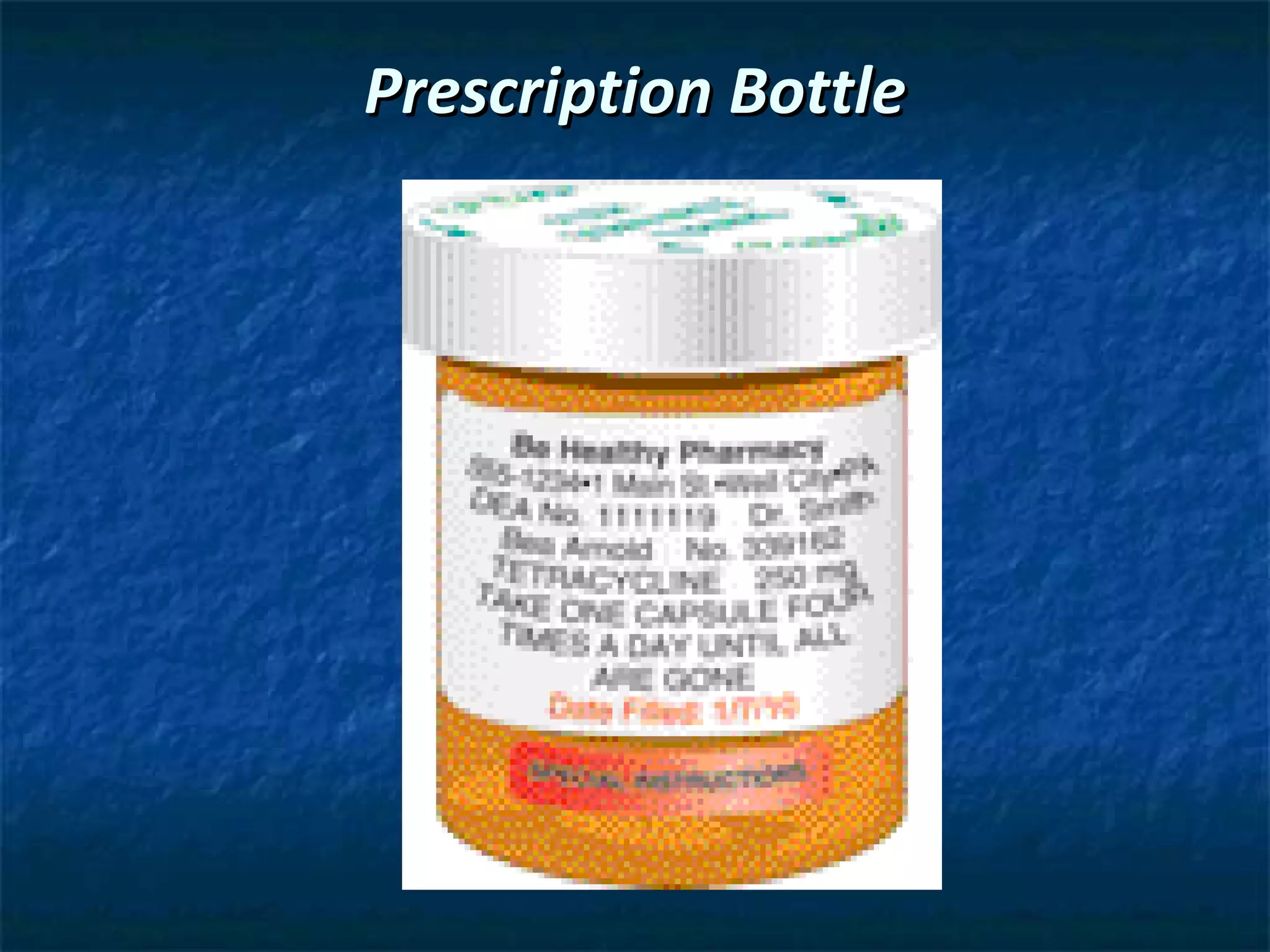



































The document outlines the history and key pieces of legislation regulating drugs in the United States, beginning with the 1906 Pure Food and Drug Act which aimed to prohibit misbranded and adulterated drugs. Major laws included the 1938 Federal Food, Drug, and Cosmetic Act which gave the FDA power to ensure drug safety, the 1970 Controlled Substances Act which classified drugs into schedules based on abuse potential, and the 1996 Health Insurance Portability and Accountability Act (HIPAA) which provided privacy protections for patient health information. The legislation established the FDA and DEA and granted them authority to approve, schedule, and regulate drugs.



































































