Unlocking Bioinformatics
with FASTATools
This presentation will guide you through FASTA tools. We will explore
the format, its importance, and common tools. We'll also cover
applications in genomics, proteomics, and transcriptomics.
SM
by SANJEEVI M
2.
Demystifying the FASTA
Format
1Structure
A FASTA file consists of a header and sequence.
2 Sequence Types
Common sequence types include DNA, RNA, and protein.
3 Codes
It uses IUPAC codes for nucleotides and amino acids.
4 Guidelines
Refer to NCBI guidelines for valid formats.
3.
Popular FASTA Tools
SequenceAlignment
BLAST and ClustalW are
used for alignment.
Sequence
Manipulation
Seqkit handles FASTA/FASTQ
manipulation.
Sequence Analysis
EMBOSS and Biopython are
used for analysis.
Sequence Conversion
Online tools and command-
line utilities handle
conversion.
4.
Sequence Alignment withBLAST
Input Sequence
Input your FASTA sequence
to NCBI BLAST.
Select Database
Choose an appropriate
database.
Run BLAST
Execute the alignment.
Interpret Results
Analyze E-value, identity,
and coverage.
5.
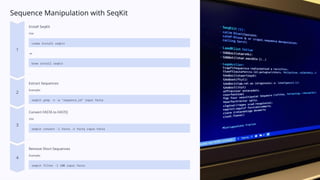
Sequence Manipulation withSeqKit
Install SeqKit
Use
conda install seqkit
or
brew install seqkit
.
Extract Sequences
Example:
seqkit grep -n -w "sequence_id" input.fasta
Convert FASTA to FASTQ
Use
seqkit convert -i fasta -o fastq input.fasta
.
Remove Short Sequences
Example:
seqkit filter -l 100 input.fasta
6.
Sequence Analysis withBiopython
Install Biopython
pip install biopython 1
Read FASTA Files
from Bio import SeqIO; for
record in
SeqIO.parse("input.fasta",
"fasta"): print(record.id,
len(record.seq))
2
Calculate Metrics
Find sequence length and GC content.
3
Find Motifs
from Bio import motifs;
instances =
motifs.create([sequence1,
sequence2]);
print(instances.search(your_
sequence))
4
7.
Best Practices andTips
Validate Files
Use online validators or command-line tools.
Handle Large Files
Consider memory and indexing.
Document Sources
Track sequence sources and modifications.
Naming Conventions
Use consistent IDs.
8.
Conclusion
FASTA tools arevital in bioinformatics. They enable sequence analysis,
alignment, and manipulation. New tools are continuously being
developed. Explore Biostars, SeqAnswers, and Biopython
documentation for more.





![Sequence Analysis with Biopython
Install Biopython
pip install biopython 1
Read FASTA Files
from Bio import SeqIO; for
record in
SeqIO.parse("input.fasta",
"fasta"): print(record.id,
len(record.seq))
2
Calculate Metrics
Find sequence length and GC content.
3
Find Motifs
from Bio import motifs;
instances =
motifs.create([sequence1,
sequence2]);
print(instances.search(your_
sequence))
4](https://image.slidesharecdn.com/unlocking-bioinformatics-with-fasta-tools-250419084854-39a12249/85/Unlocking-Bioinformatics-with-FASTA-Tools-pptx-6-320.jpg)























































