Download to read offline


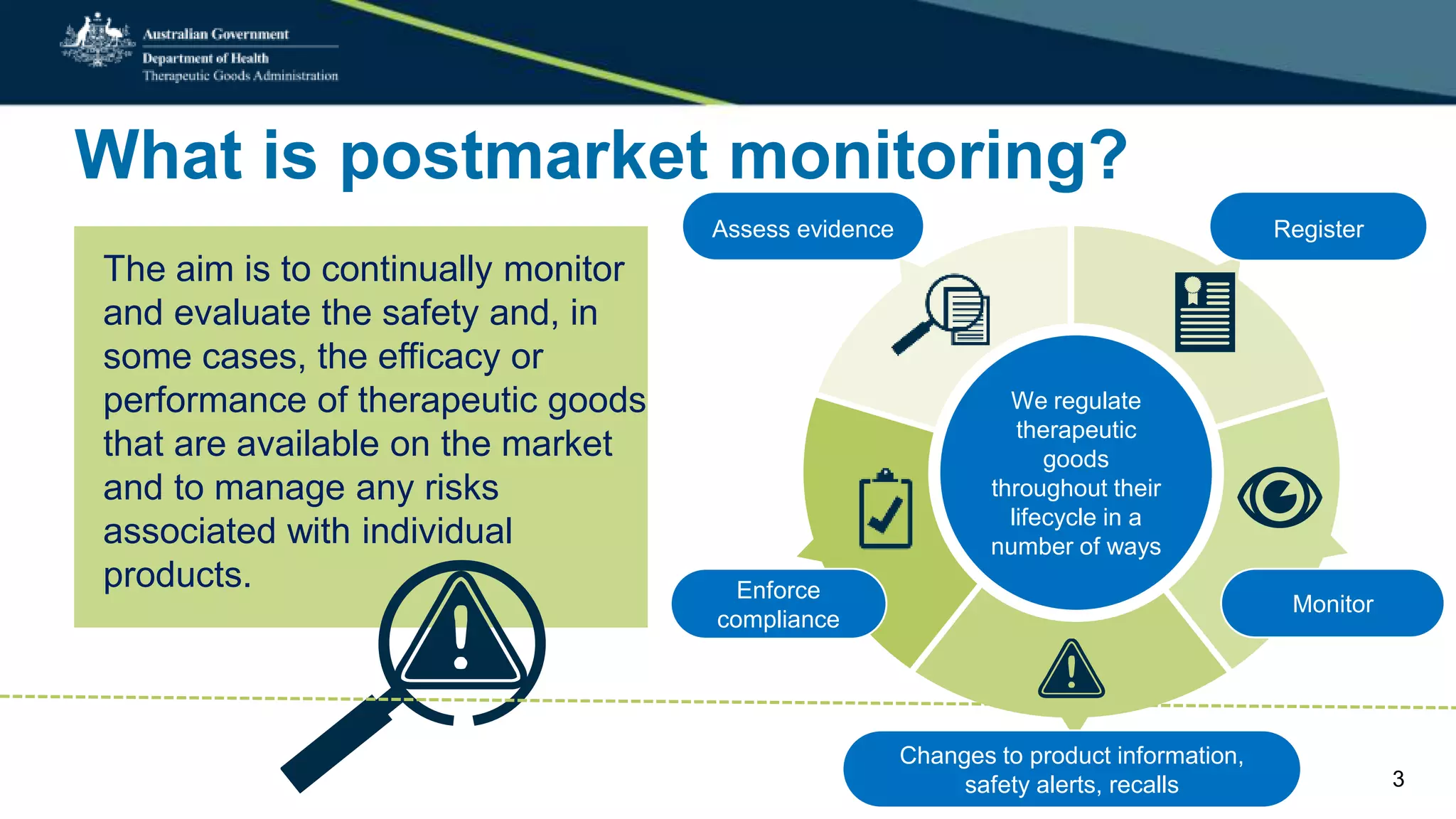


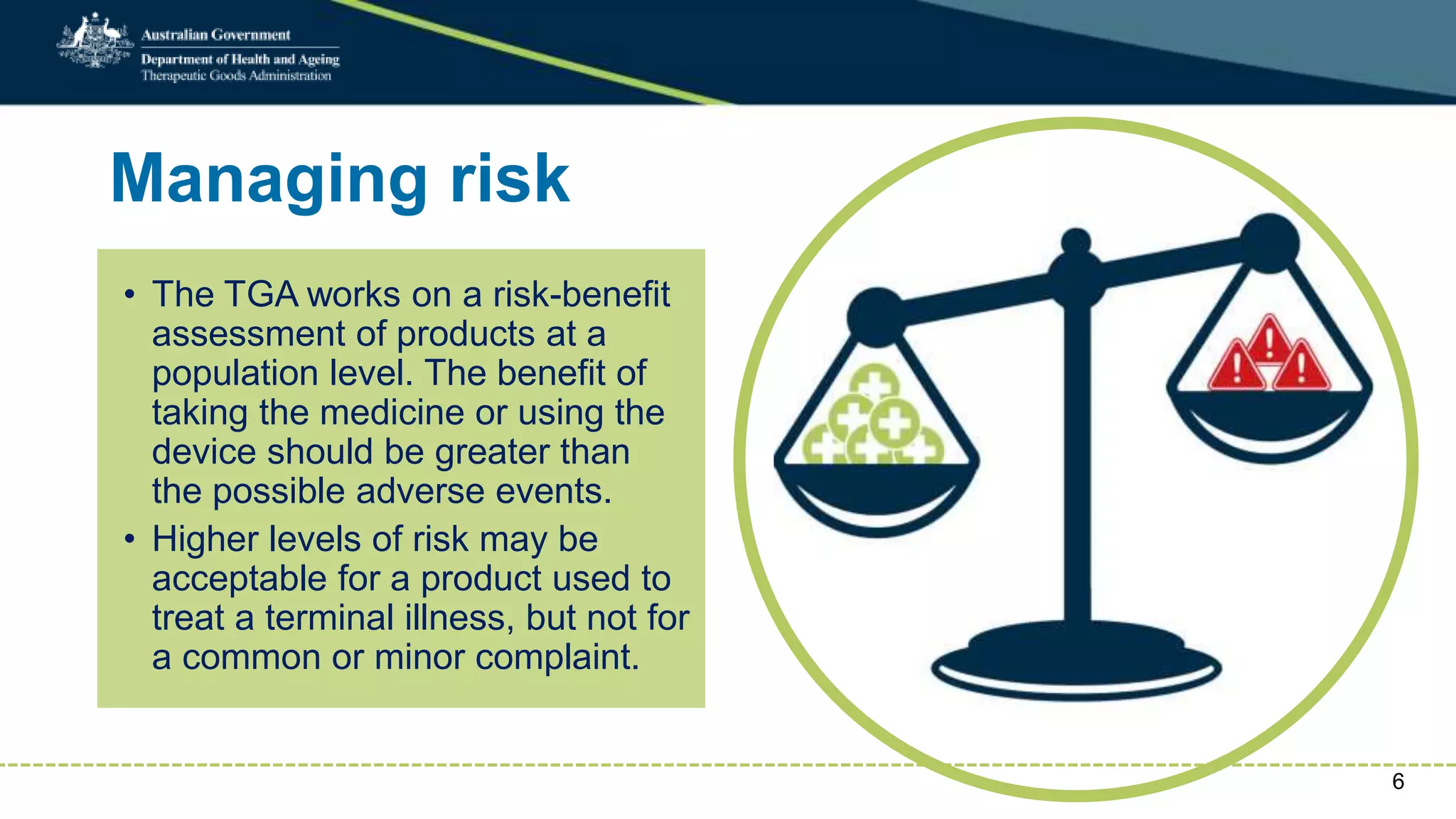



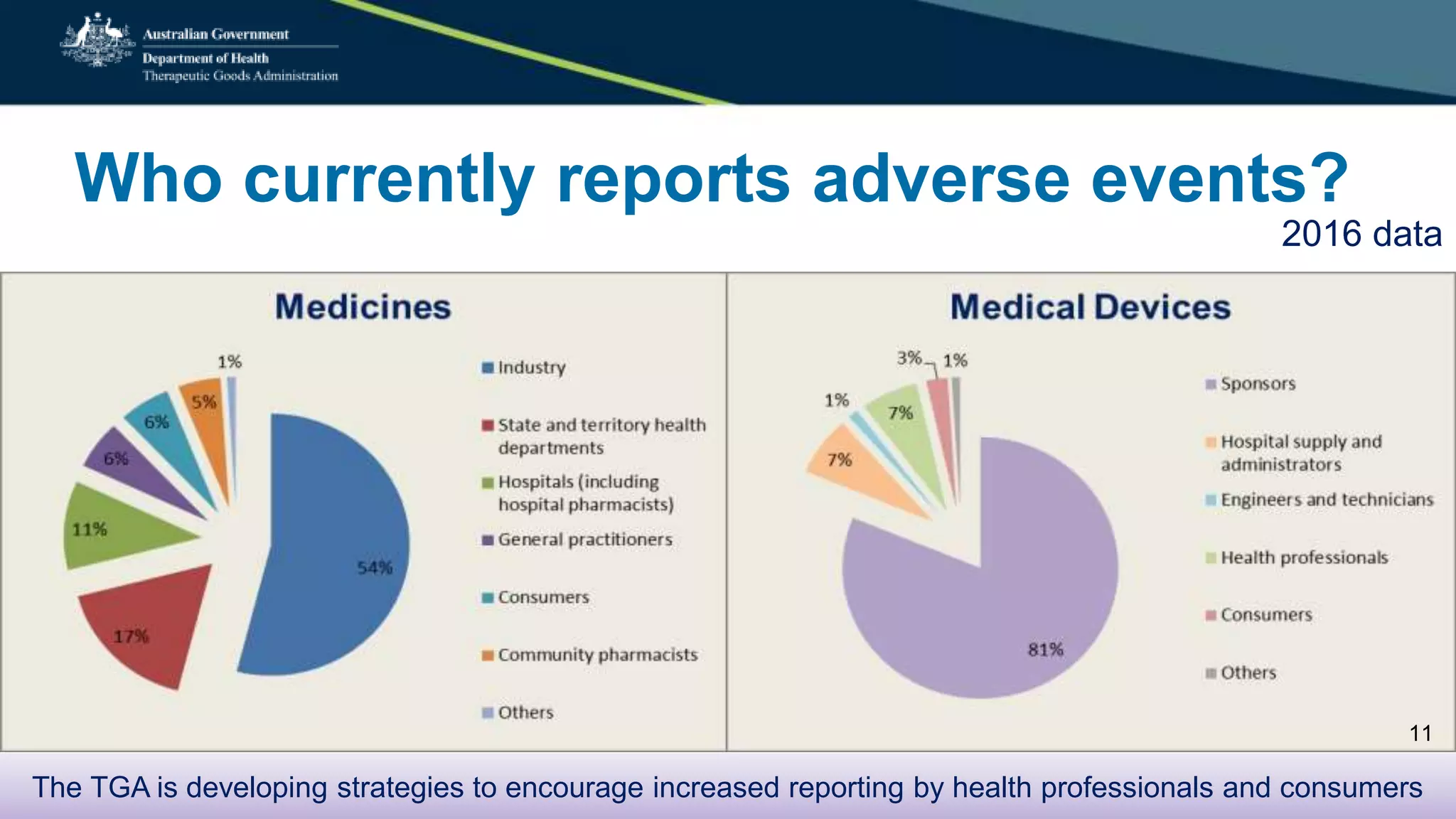













Postmarket monitoring involves the continuous evaluation of the safety and efficacy of therapeutic goods available in the market, aiming to manage associated risks. It is crucial as some risks, such as those related to Vioxx, become apparent only after product registration; this led to enhanced monitoring by regulators globally. The monitoring process includes tools like risk management plans and adverse event reporting, with health professionals playing a key role in reporting any suspected adverse events.























































































