Downloaded 19 times




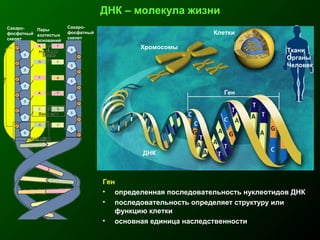


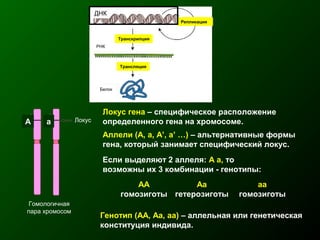
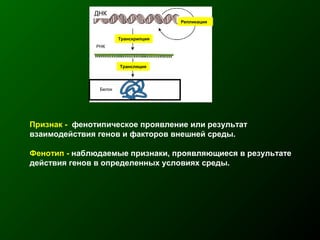

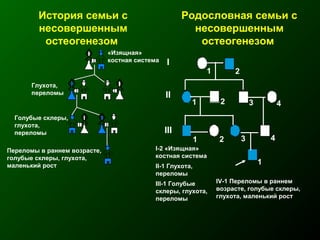


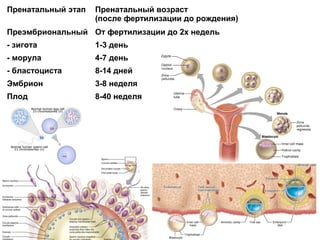







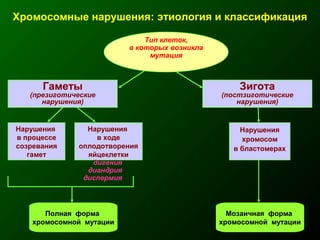














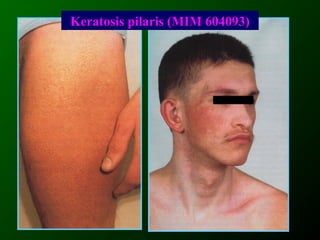






























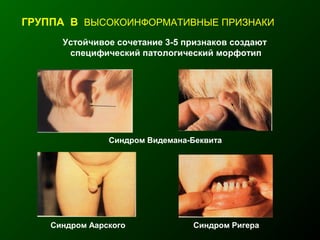


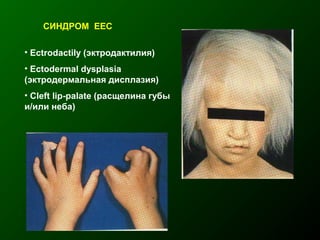


Документ освещает основы человеческого развития через призму генетики, акцентируя внимание на механизмах функционирования клеток и эволюции. Рассматриваются различные генетические аспекты, включая медицинскую генетику и наследственные болезни, а также этапы эмбрионального и постнатального развития. Включены статистические данные по наследственным заболеваниям и описаны характерные синдромы с хромосомными аномалиями.





















































