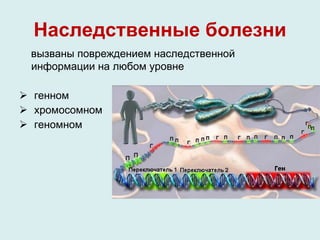
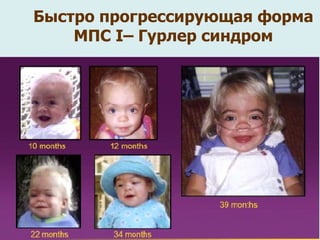
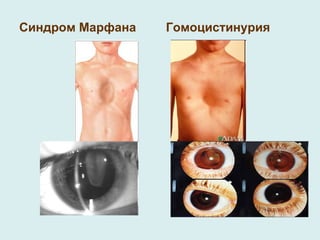
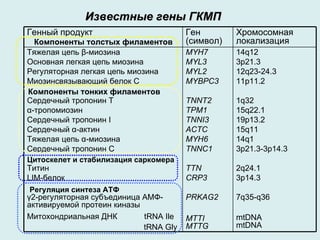
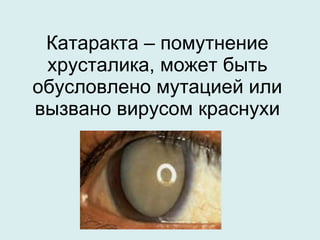
Документ рассматривает гетерогенность наследственных заболеваний, их диагностику и лечение. Упоминаются различные типы генетических мутаций, приводящих к заболеваниям, а также важность раннего выявления и консультирования специалистов. Генетическая гетерогенность обуславливает сложности в клиническом полиморфизме и диагностике наследственных болезней.