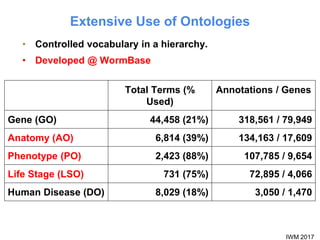
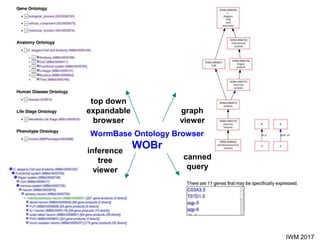
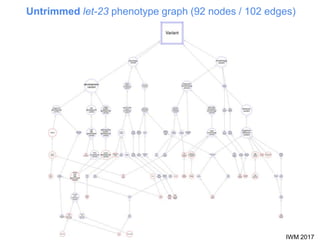
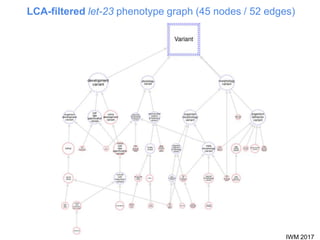
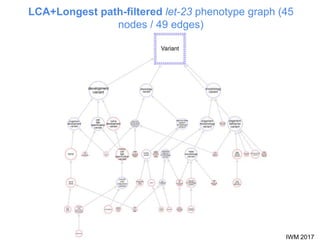
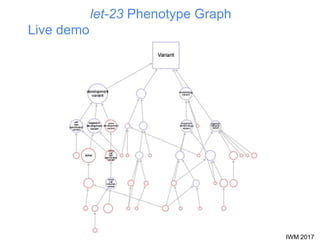
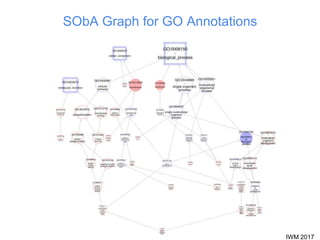
This document describes SObA, a tool that generates concise graphical summaries of ontology-based annotations for genes. It summarizes extensive use of ontologies by WormBase to annotate C. elegans genes with terms related to gene function, anatomy, phenotypes, life stages and human diseases. It addresses the problem that lists of mutant phenotypes for genes with many annotations are difficult to comprehend. SObA generates interactive graphs that integrate the ontology hierarchy and logical inferences to provide a complete yet simplified view of a gene's most essential ontology annotations. It was shown to better represent the biological meaning than untrimmed lists of annotations.

































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)


![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)











