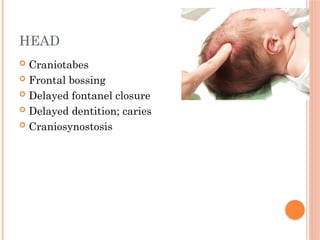
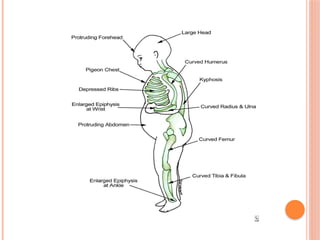
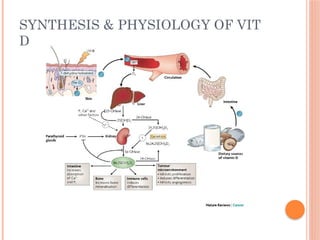
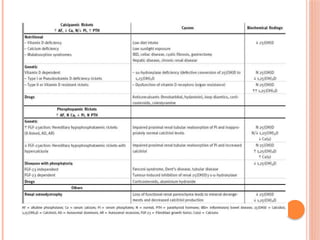
The document provides a comprehensive overview of rickets, detailing various causes such as vitamin D disorders, calcium and phosphorus deficiencies, and renal losses. It outlines clinical features, diagnosis, treatment options, prognosis, and prevention strategies, emphasizing the importance of adequate vitamin D, calcium, and phosphorus intake. Specific types of vitamin D-dependent rickets and their treatments are also discussed, along with genetic aspects and related complications.






























![TREATMENT
providing adequate calcium, typically as a
dietary supplement
700 mg/day of elemental calcium [1-3 yr age],
1,000 mg/day of elemental calcium [4-8 yr age],
1,300 mg/day of elemental calcium [9-18 yr age]
Vitamin D supplementation is necessary if there
is concurrent vitamin D deficiency .
Prevention
discouraging early cessation of breast-feeding
and increasing dietary sources of calcium
school-based milk programs](https://image.slidesharecdn.com/rickets-250117131810-ed10d4b8/85/RICKETS-types-clinical-features-and-management-33-320.jpg)






















































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)











