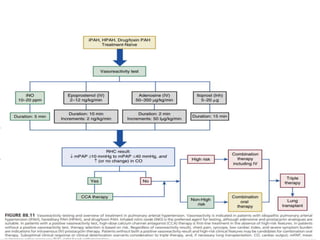
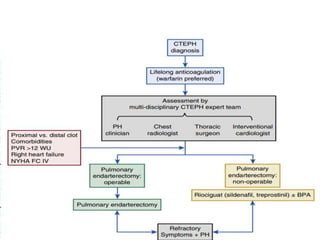
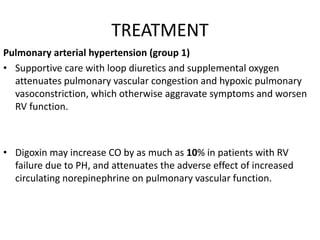
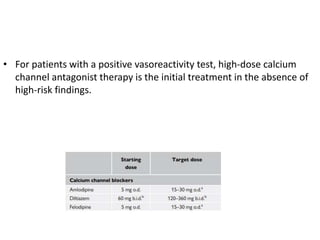
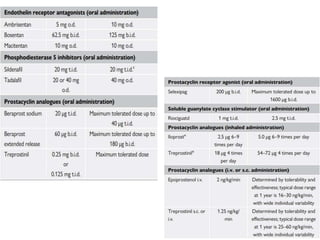
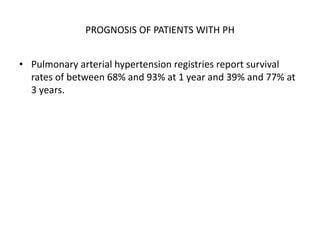
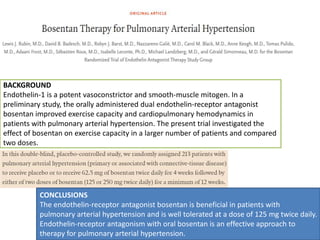
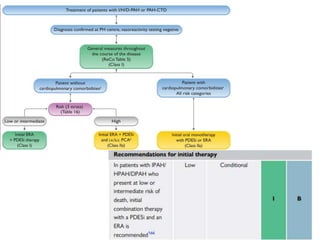
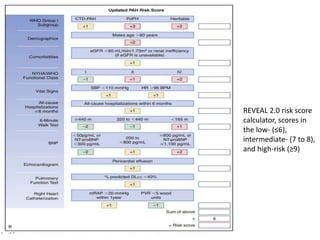
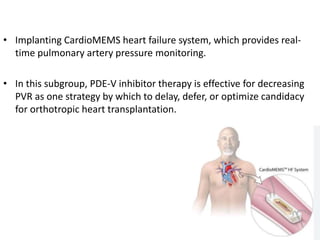
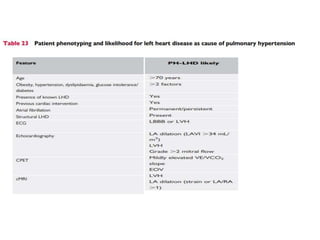
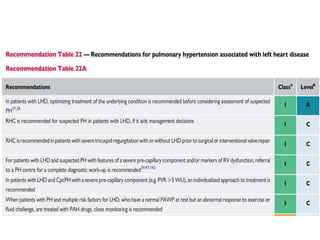
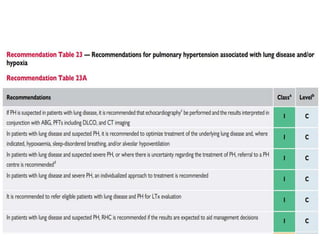
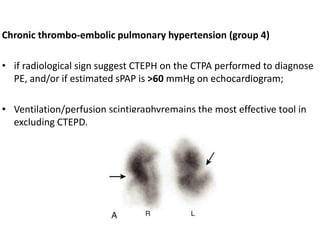
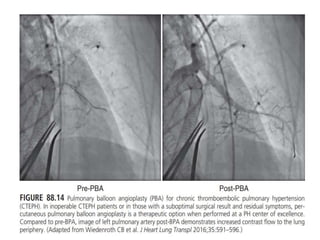
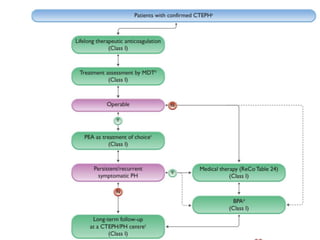
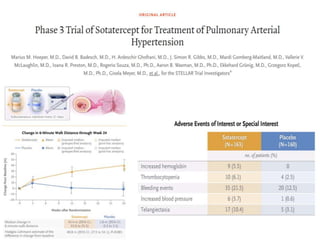
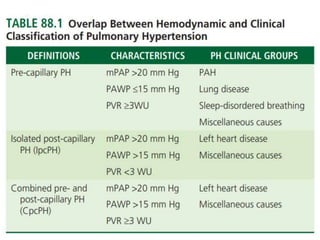
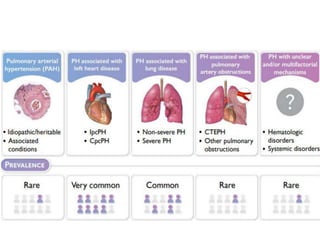
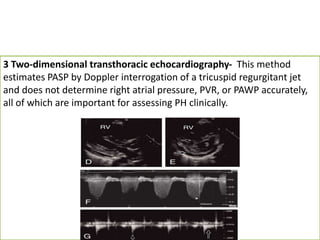
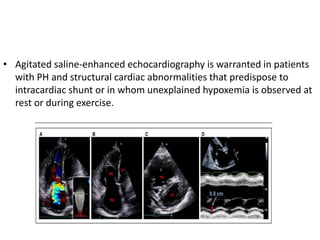
The document outlines the diagnosis and management of pulmonary hypertension (PH), detailing its definition, initial assessments, and various diagnostic tools. It emphasizes the importance of an integrated diagnostic approach, including imaging, functional tests, and cardiac catheterization, alongside treatment options depending on the etiology of PH. Additionally, it discusses recent studies evaluating therapies such as bosentan and riociguat, highlighting their safety and efficacy in treating pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension.







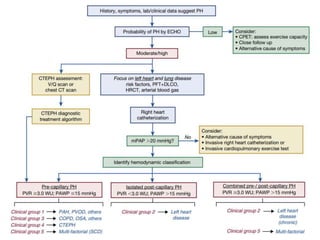
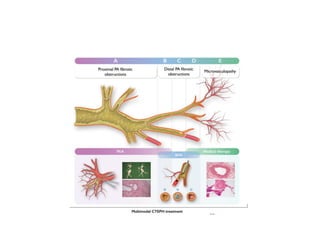
![5 Computed tomographic chest imaging and nuclear
ventilation/perfusion (V/Q) scintigraphy- Mismatched perfusion
defects on ventilation-perfusion (V/Q) scintigraphy is highly suggestive
of CTEPH.
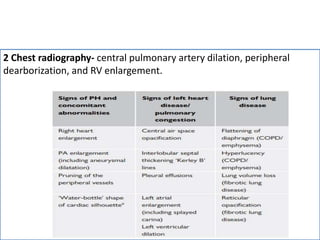
• The CT signs suggesting the presence of PH include an enlarged PA
diameter, a PA-to-aorta ratio >0.9, and enlarged right heart
chambers.
• A combination of three parameters (PA diameter ≥30 mm, RVOT wall
thickness ≥6 mm, and septal deviation ≥140° [or RV:LV ratio ≥1]) is
highly predictive of PH.](https://image.slidesharecdn.com/pulmonaryhypertension-240131132156-8513b691/85/Pulmonary-Hypertension-pptx-15-320.jpg)





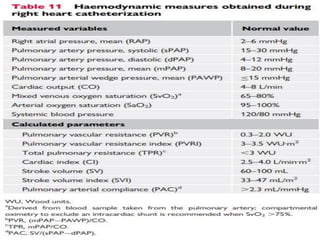
![• Pulmonary vascular resistance ([mPAP−PAWP]/CO) should be
calculated for each patient.
• All pressure measurements, including PAWP, should be performed at
end expiration (without breath-holding manoeuvre).
• In patients with large intrathoracic pressure changes during the
respiratory cycle (i.e. COPD, obesity, during exercise), it is appropriate
to average over at least three to four respiratory cycles.](https://image.slidesharecdn.com/pulmonaryhypertension-240131132156-8513b691/85/Pulmonary-Hypertension-pptx-21-320.jpg)