Protocols for Immunoassay. Procedure for different types of immunoassays.
Radioimmunoassay
ELISA and it's types with examples.
Immunofluorescence
Chemiluminescence immunoassay
Immunochromatographic assay
Immunoprecipitation
Western blotting
2
Immunoassays
An immunoassayis a biochemical test that measures the
presence or concentration of a substance, often in biological
samples, by using an antibody or antigen reaction.
Immunoassays rely on the specificity of antibodies to bind
to unique molecules, called antigens, allowing for sensitive
detection of various targets such as proteins, hormones,
viruses, and drugs.
They are widely used in medical diagnostics, research, and
pharmacology for applications like detecting infections,
measuring hormone levels, and monitoring therapeutic
drugs.
4
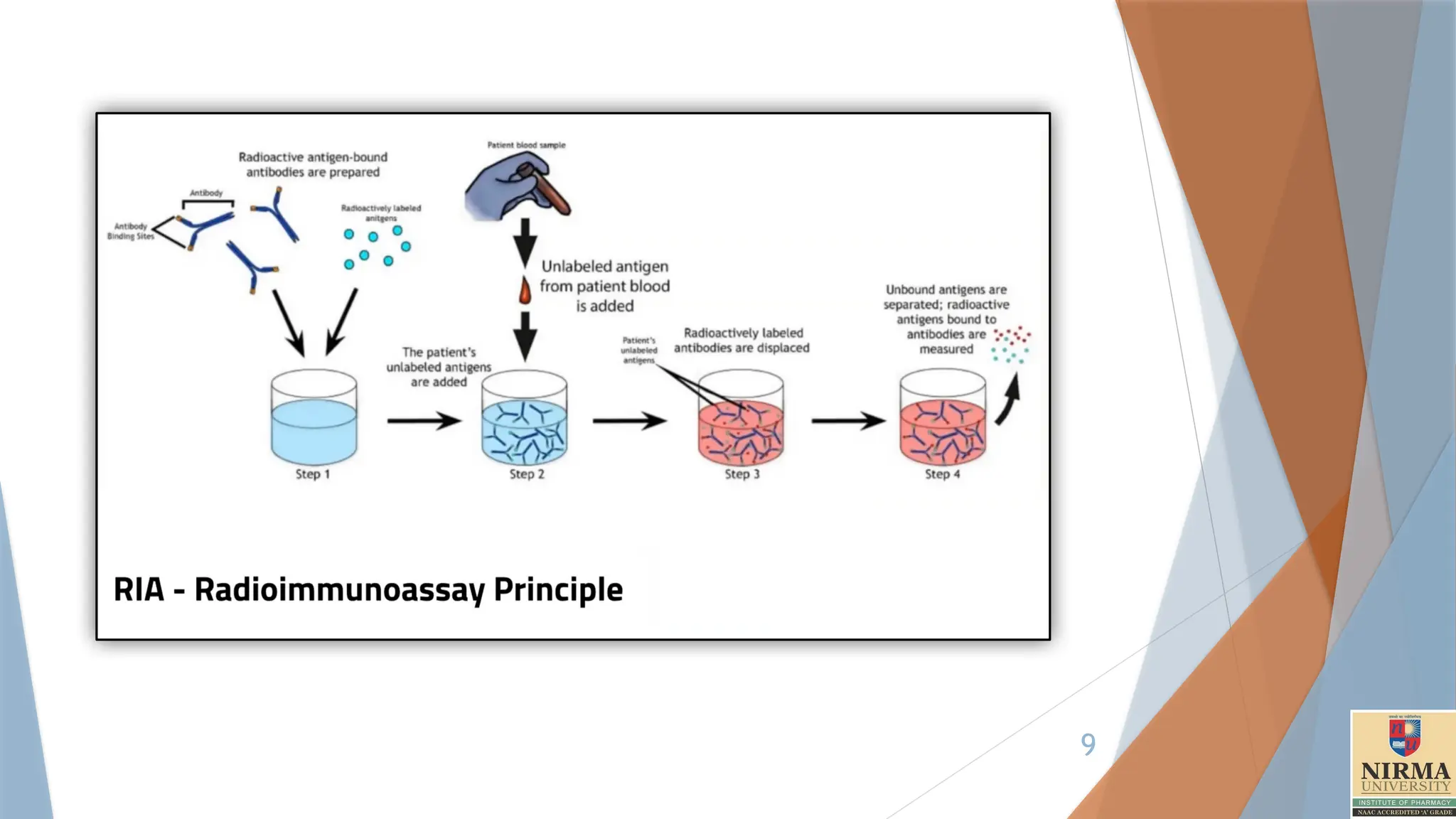
1. Radioimmunoassay
Radioimmunoassay(RIA) is a sensitive laboratory technique used to
measure trace amounts of substances, typically hormones, drugs, or
proteins, in a sample. It combines principles of immunology with
radioactivity, utilizing antibodies to detect and quantify specific molecules.
Developed by Yalow and Berson. Because the method was so precise,
they were able to prove that type 2 diabetes is caused by the body's
inefficient use of insulin. Previously it was thought that the disease was
caused by a lack of insulin.
Principle of
Radioimmunoassay
RIA operates on the principle of competitive binding, where a known
amount of a radioactively-labelled insulin (the “radioligand”)
competes with the unlabelled insulin in the sample for binding sites on
a specific antibody.
Since the radioligand and the target molecule (insulin) share the same
antibody binding sites, the amount of radioactivity measured after
equilibrium reflects the concentration of the unlabelled insulin.
5.
5
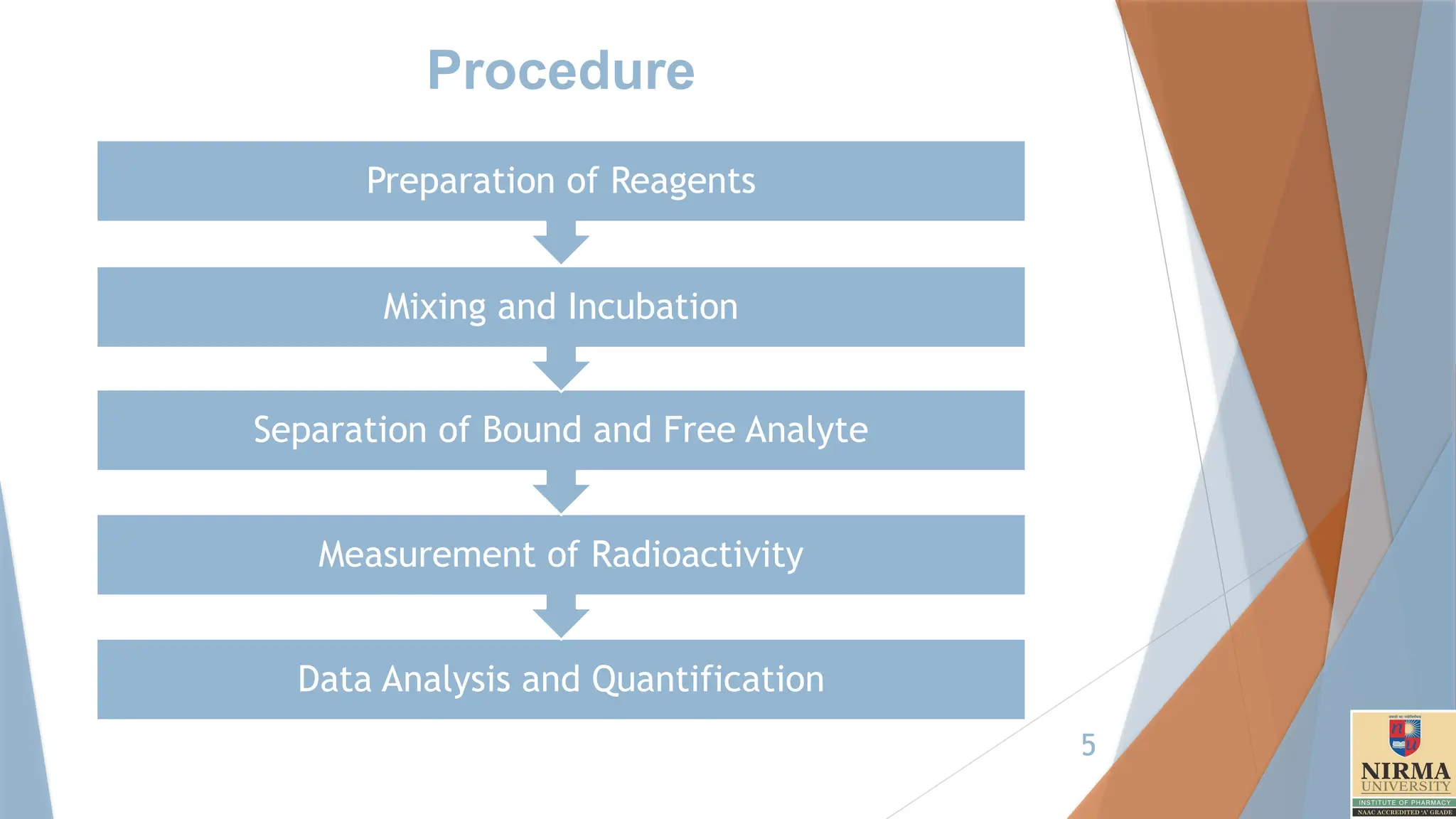
Procedure
Data Analysis andQuantification
Measurement of Radioactivity
Separation of Bound and Free Analyte
Mixing and Incubation
Preparation of Reagents
6.
6
1. Preparation ofReagents:
A known quantity of the substance to be measured (Insulin) is
labelled with a radioactive isotope, often iodine-125 or tritium.
The antibody Anti-insulin antibodies (specific for human insulin)
specific to the analyte is prepared, ensuring high affinity and
specificity.
Create a series of standard insulin concentrations to establish the
standard curve. (e.g., 0, 2.5, 5, 10, 20, 40 µU/mL).
2. Mixing and Incubation:
A small amount of the radiolabelled insulin and the unlabelled
sample insulin are mixed with the antibody in a test tube or well.
During incubation, the labelled and unlabelled insulin compete to
bind to the limited antibody binding sites.
The mixture is left to incubate for 8-12 hours to reach binding
equilibrium.
7.
7
3. Separation ofBound and Free Analyte:
After incubation, it is necessary to separate the bound insulin-
antibody complex from the free (unbound) insulin.
This can be done by washing them with buffer solution of (A) -
Trifluoroacetic acid and (B) - 60% Acetonitrile + 1%
Trifluoroacetic acid + 39% distilled water.
The bound complex can then be isolated by centrifugation or
filtration.
4. Measurement of Radioactivity:
The radioactivity of the bound complex is measured using a
Gamma counter if iodine-125 is used or a scintillation counter for
tritium.
The amount of radioactivity in the bound fraction is inversely
proportional to the concentration of the analyte in the sample.
8.
8
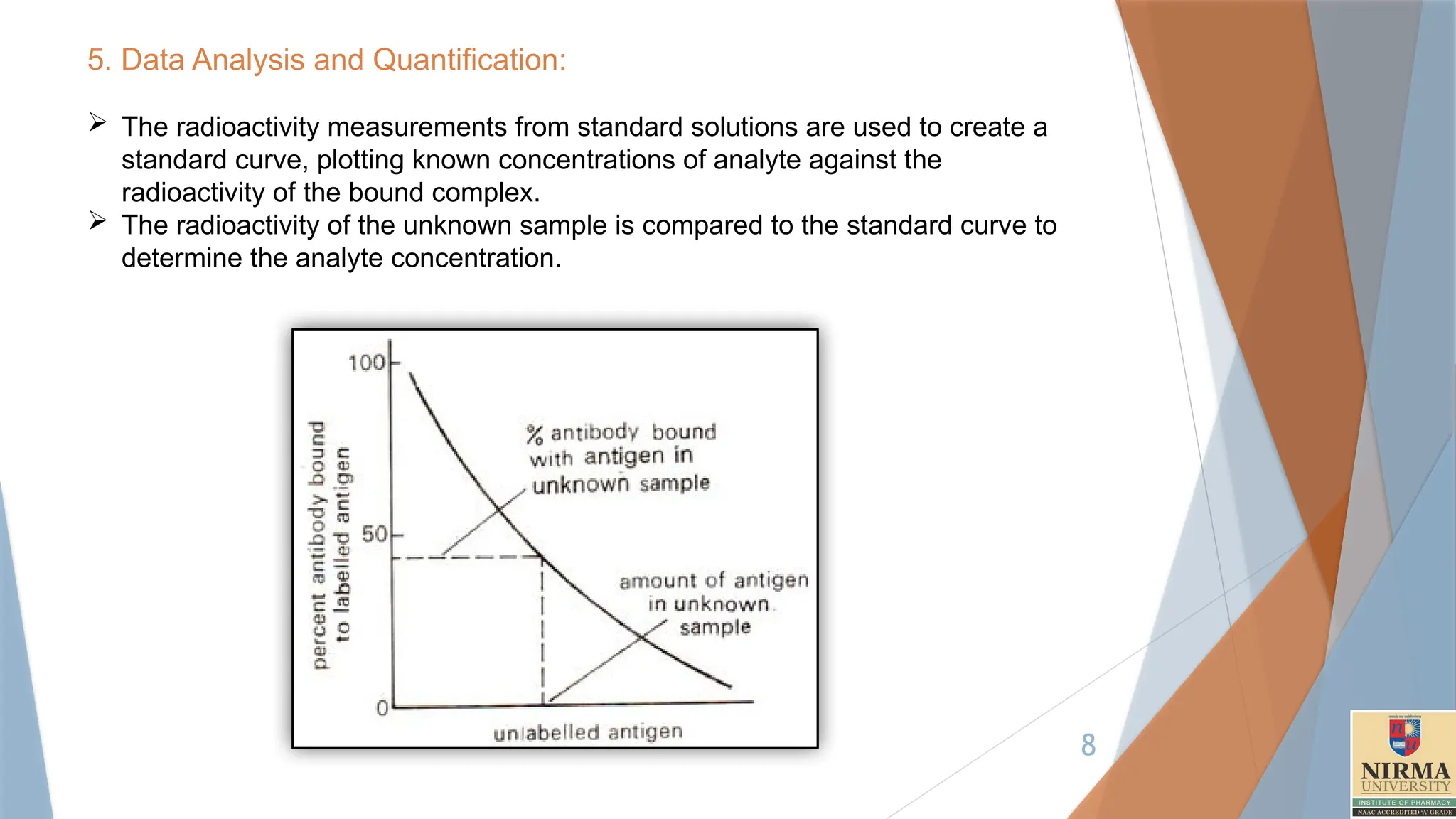
5. Data Analysisand Quantification:
The radioactivity measurements from standard solutions are used to create a
standard curve, plotting known concentrations of analyte against the
radioactivity of the bound complex.
The radioactivity of the unknown sample is compared to the standard curve to
determine the analyte concentration.
10
2. ELISA –Enzyme linked
Immunosorbent Assay
Enzyme-Linked Immunosorbent Assay (ELISA) is a commonly used
laboratory technique for detecting and quantifying specific proteins,
antibodies, or antigens in a sample.
The principle of ELISA relies on antibody-antigen interactions, and it
involves using an enzyme-labelled antibody or antigen that produces a
measurable signal, often a colour change.
12
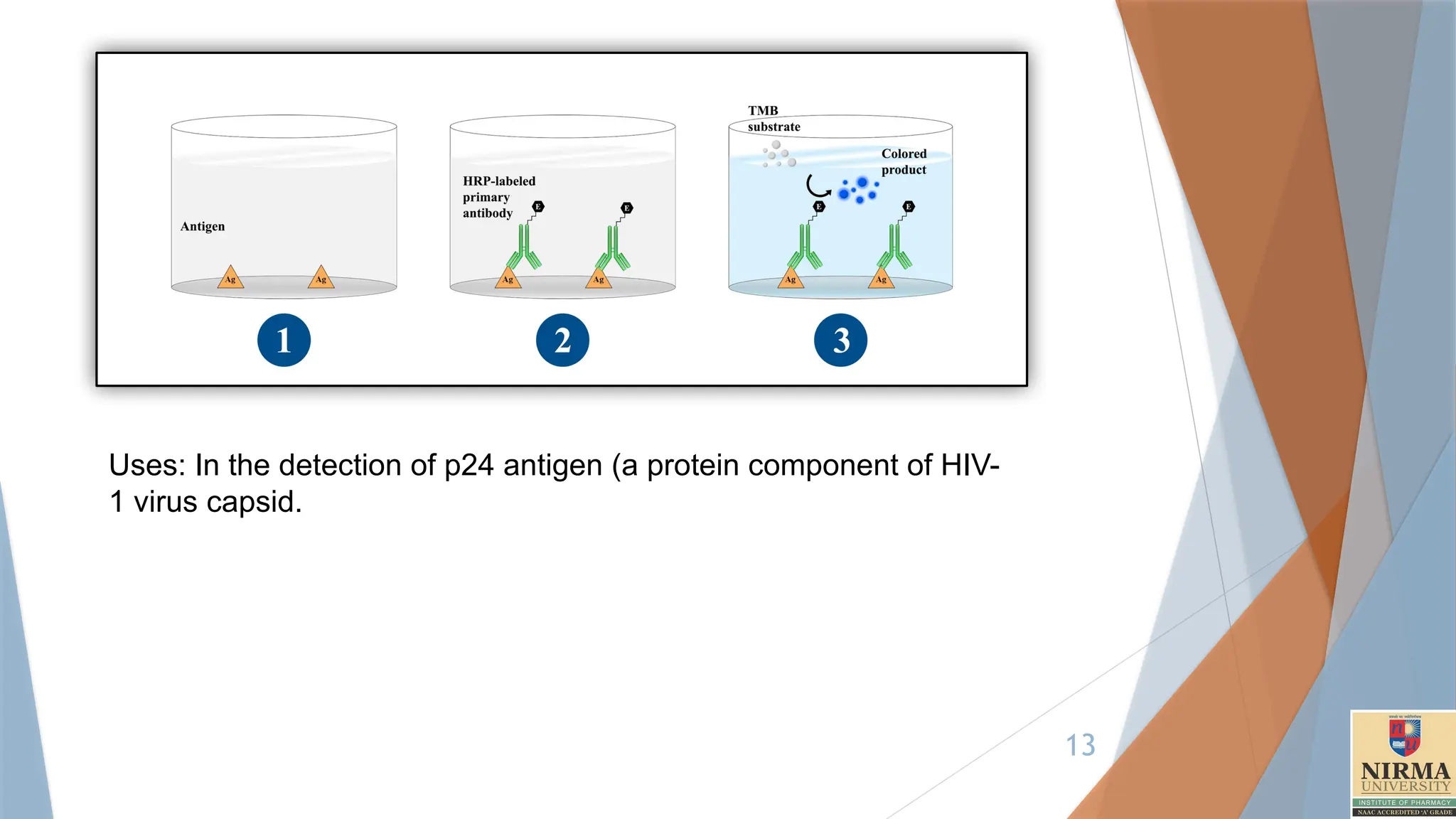
1. Direct ELISA
Principle:In direct ELISA, the antigen is first immobilized on the plate, and
then a single enzyme-labelled (conjugated) antibody specific to the target
antigen is added to detect it.
Steps:
Coating: The antigen is attached directly to the surface of the microplate
well(96 Well plate).
Blocking: A blocking buffer (e.g., BSA, skim milk, Tween-20) is used to
prevent nonspecific binding. Incubate for at least two hours at room
temperature or overnight at 4°C. Wash the plate three or more times
with a washing buffer(PBS).
Detection: An enzyme-conjugated antibody that binds directly to the
target antigen is added.
Substrate Addition: The enzyme substrate is added, producing a colour
change if the target antigen is present.
Measurement: The colour intensity is measured, which is proportional to
the antigen concentration.
13.
13
Uses: In thedetection of p24 antigen (a protein component of HIV-
1 virus capsid.
14.
14
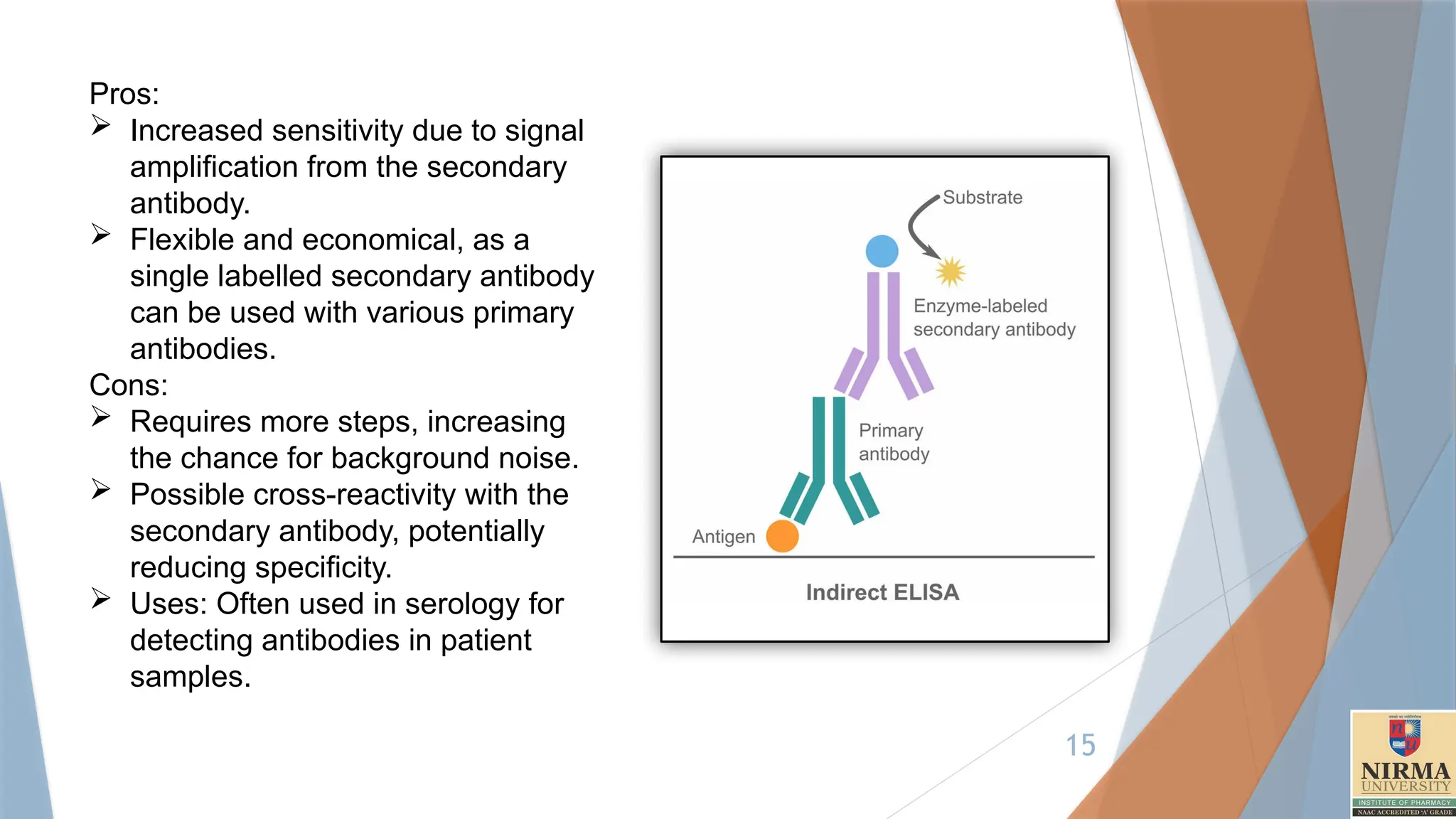
2. Indirect ELISA
Principle:In indirect ELISA, an unconjugated primary antibody specific to
the antigen is used, followed by an enzyme-conjugated secondary
antibody that binds to the primary antibody.
Steps are as follows:
Coating: The antigen is immobilized on the plate surface.
Blocking: Blocking solution (e.g., BSA, skim milk, Tween-20) is added
to prevent nonspecific binding.
Primary Antibody: An antibody specific to the target antigen is added
Secondary Antibody: An enzyme-linked secondary antibody that binds
to the primary antibody is added
Substrate Addition: The enzyme substrate is added, producing a colour
change if the target antigen is present.
Measurement: The colour intensity correlates with the amount of
antigen.(Proportional).Detection by colorimetry.
15.
15
Pros:
Increased sensitivitydue to signal
amplification from the secondary
antibody.
Flexible and economical, as a
single labelled secondary antibody
can be used with various primary
antibodies.
Cons:
Requires more steps, increasing
the chance for background noise.
Possible cross-reactivity with the
secondary antibody, potentially
reducing specificity.
Uses: Often used in serology for
detecting antibodies in patient
samples.
16.
16
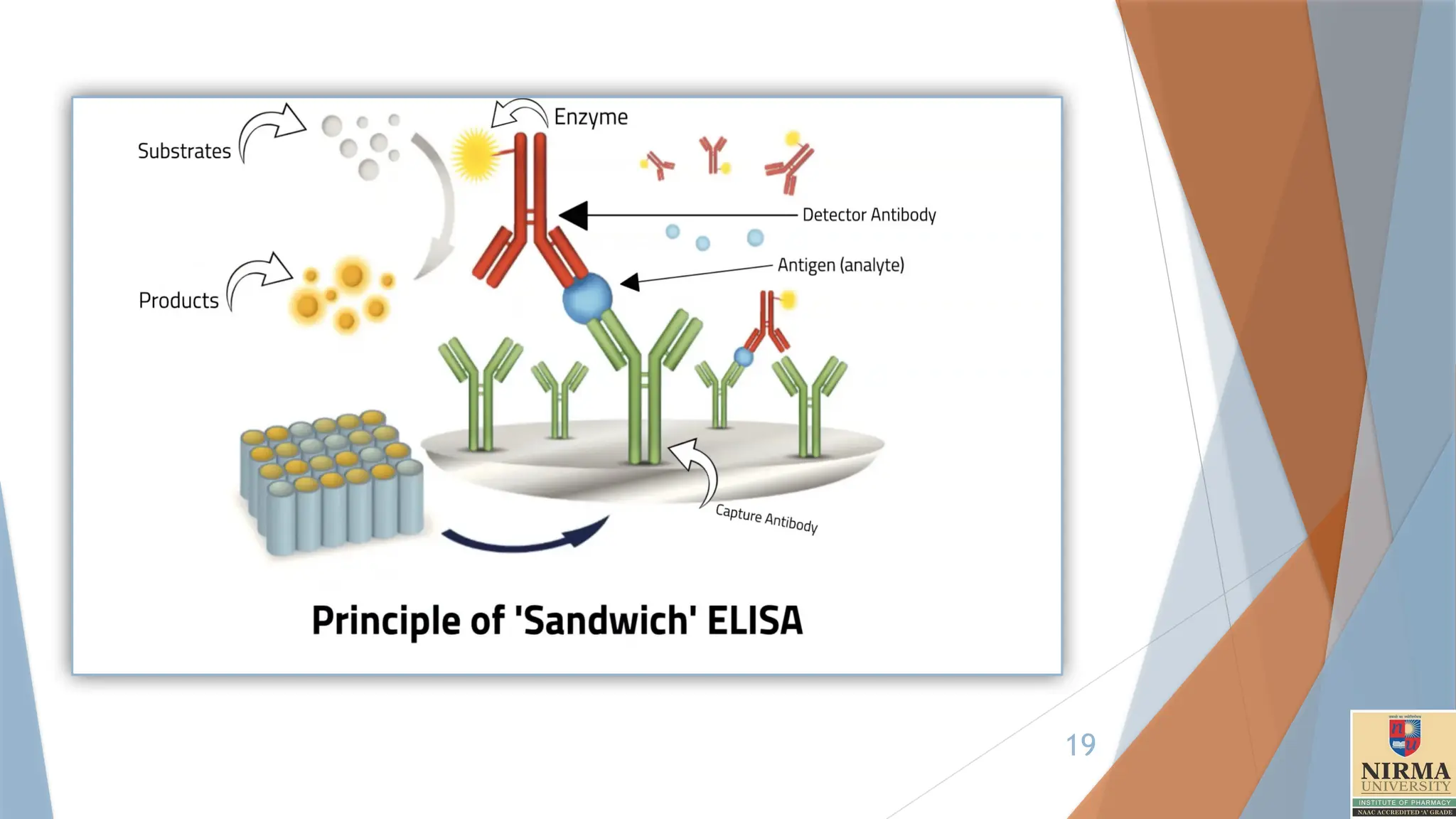
3. Sandwich ELISA
Principle:Sandwich ELISA uses two different antibodies (capture and
detection antibodies) that bind to different sites on the target antigen,
"sandwiching" it between them. This type is highly specific and sensitive.
Steps are as follows:
1. Coating the Plate:
A 96-well microtiter plate is coated with a capture antibody (Anti-p24
antibody) specific to the target HIV antigen (e.g., p24 antigen).
It is generally recommended to use affinity-purified antibodies for optimal
signal-to-noise ratio.
17.
17
The plateis incubated (8-10 Hours) to allow the antibody to adhere to
the surface of the wells.
Excess antibody is washed away, and unbound sites on the plate are
blocked using a blocking agent (e.g., bovine serum albumin-BSA or Skim
milk) to prevent nonspecific binding.
2. Sample Addition:
The patient's blood or plasma sample is added to the wells. If the HIV
antigen is present, it will bind to the immobilized capture antibody.
The plate is incubated to allow sufficient binding time (1-2 hours at room
temp.), and unbound material is washed away.
3. Addition of Detection Antibody:
A secondary antibody, labelled with an enzyme (e.g., horseradish
peroxidase [HRP]), is added to the wells. This detection antibody binds to
a different epitope of the HIV antigen, completing the "sandwich."
Unbound detection antibody is washed away after incubation.
18.
18
4. Enzyme-Substrate Reaction:
A substrate for the enzyme (e.g., TMB [3,3’,5,5'-tetramethy|benzidine]) is
added. The enzyme catalyses a reaction that produces a colour change.
The intensity of the colour is directly proportional to the amount of HIV
antigen in the sample.
5. Measurement:
The colour intensity is measured using a spectrophotometer at a specific
wavelength (e.g., 450 nm).
A standard curve is used to quantify the antigen concentration, if needed.
Applications of HIV/AIDS Diagnosis:
Sandwich ELISA is widely used in combination with other tests ( e.g.
antibody detection ELISA, Western Blot, or PCR) to confirm HIV status. It
plays a crucial role in early detection, monitoring disease progression, and
evaluating the effectiveness of Antiretroviral therapy (ART).
20
4. Competitive ELISA
Itoperates on the principle of competition between a target analyte in the
sample and a labelled analyte for a limited number of binding sites on a
specific antibody.
Example of a competitive ELISA is the detection of aflatoxins in food
products. Aflatoxins are toxic secondary metabolites produced by certain
fungi and pose a significant health risk.
Key Steps in Competitive ELISA
1.Coating the Plate:
The wells of the microtiter plate are pre-coated with an anti-aflatoxin
antibody.
1–2 hours at 37°C, or overnight at 4°C.
2. Blocking
Blocking agents (e.g. casein or BSA) are added to block non-specific
binding sites. (30 minutes to 1 hour at room temperature (RT) or
37°C)
21.
21
3. Adding sampleand labelled aflatoxin:
A fixed amount of enzyme-labelled aflatoxin (e.g., aflatoxin conjugated
with HRP) is mixed with the food extract containing an unknown
concentration of aflatoxin.
This mixture is added to the wells, where the unlabelled aflatoxin (from
the sample) competes with the labelled aflatoxin for the limited binding
sites of the antibody.
4. Incubation Time: 1–2 hours at RT or 37°C.
The plate is incubated to allow the competition to occur.
Higher concentrations of aflatoxin in the food sample will result in less
binding of the labelled aflatoxin.
5. Washing:
Unbound labelled aflatoxin and other impurities are washed away by
PBS (Phosphate Buffer Saline) to ensure only the antigen-antibody
complexes remain.
(PBS): Contains NaCl, KCl, Na PO , and KH PO .
₂ ₄ ₂ ₄
22.
22
6. Substrate Addition:
A substrate for the enzyme (e.g., TMB- 3,3′,5,5′-Tetramethylbenzidine.) is
added. The HRP enzyme catalyses a colour change in the substrate.(blue
colour)
The intensity of the colour is inversely proportional to the aflatoxin
concentration in the sample (more aflatoxin in the sample = weaker colour).
7. Measurement:
The colour intensity is measured using a spectrophotometer at 450 nm.
A standard curve is generated using known aflatoxin concentrations, and the
aflatoxin concentration in the food sample is determined by interpolation.
23.
23
3. Immunofluorescence
Fluorescentimmunoassay (FIA) is an analytical technique that
combines immunology and fluorescence detection to measure
specific antigens or antibodies within a sample.
This method is widely used in clinical diagnostics, research, and
pharmaceutical applications due to its sensitivity, specificity, and
speed.
Types of Immunofluorescence are:
24.
24
In thistechnique, known as immunofluorescence, fluorescent compounds
such as fluorescein and rhodamine are in common use, but other highly
fluorescent substances are also routinely used, such as phycoerythrin,
an intensely coloured and highly fluorescent pigment obtained from algae.
Fluorescein, an organic dye that is the most widely used label for
immunofluorescence procedures, absorbs blue light (490 nm) and emits
an intense yellow-green fluorescence (517 nm).
Rhodamine, another organic dye, absorbs in the yellow-green range
(515 nm) and emits a deep red fluorescence (546 nm).
Phycoerythrin is an efficient absorber of light (~30-fold greater than
fluorescein) and a brilliant emitter of red fluorescence, stimulating its wide
use as a label for immunofluorescence.
Example: Presence of SARS-CoV-2 nucleoprotein in infected cell
cultures using IFA.
Here the fluorescence dye used is fluorescein isothiocyanate (FITC).
Also used to detect Systemic Lupus Erythematosus (SLE) an auto
immune disease.
27
4. Chemiluminescence
Immunoassay
ChemiluminescenceImmunoassay (CLIA) is an immunoassay
technique that combines chemiluminescence (light emission from a
chemical reaction) with immunochemical reactions to detect and
measure various substances, such as hormones, proteins, and
drugs, in biological samples.
It is known for its high sensitivity and specificity, making it widely
used in clinical laboratories for diagnostic purposes.
Can also be used to detect the gp-41 antigen of AIDS virus.
Principle:
The principle of CLIA and ELISA is similar. However the enzyme
linked secondary antibody is replaced by HRP linked secondary
antibody, and chromogenic substrate is replaced by luminol and its
derivatives with electron transport agent.
28.
28
Procedure:
1. Sample preparation:collect and prepare the sample (e.g. blood serum,
plasma or other biological fluids) to be tested. If necessary, dilute the
sample or treat it to optimize the condition for antigen-antibody binding.
2. Immobilization of the antigen: an antigen specific to the target antibody
is immobilized on a solid phase, such as the walls of a microtiter,
magnetic beads, or other surfaces. In some formats, the antibody may
be immobilized instead.
3. Incubation with sample: the prepared sample is added to the
immobilized antigen, allowing the antibody present in the sample to bind
specifically to the antigen. After an appropriate incubation period,
unbound molecules are washed away to reduce non-specific binding.
4. Addition of the chemiluminescent-labelled detection agent: a
secondary antibody labelled with a chemiluminescent marker is added to
the reaction. This secondary antibody binds to the antigen or to the
primary antibody-antigen complex, depending on the assay format.
Following incubation and unbound reagents are washed off.
29.
29
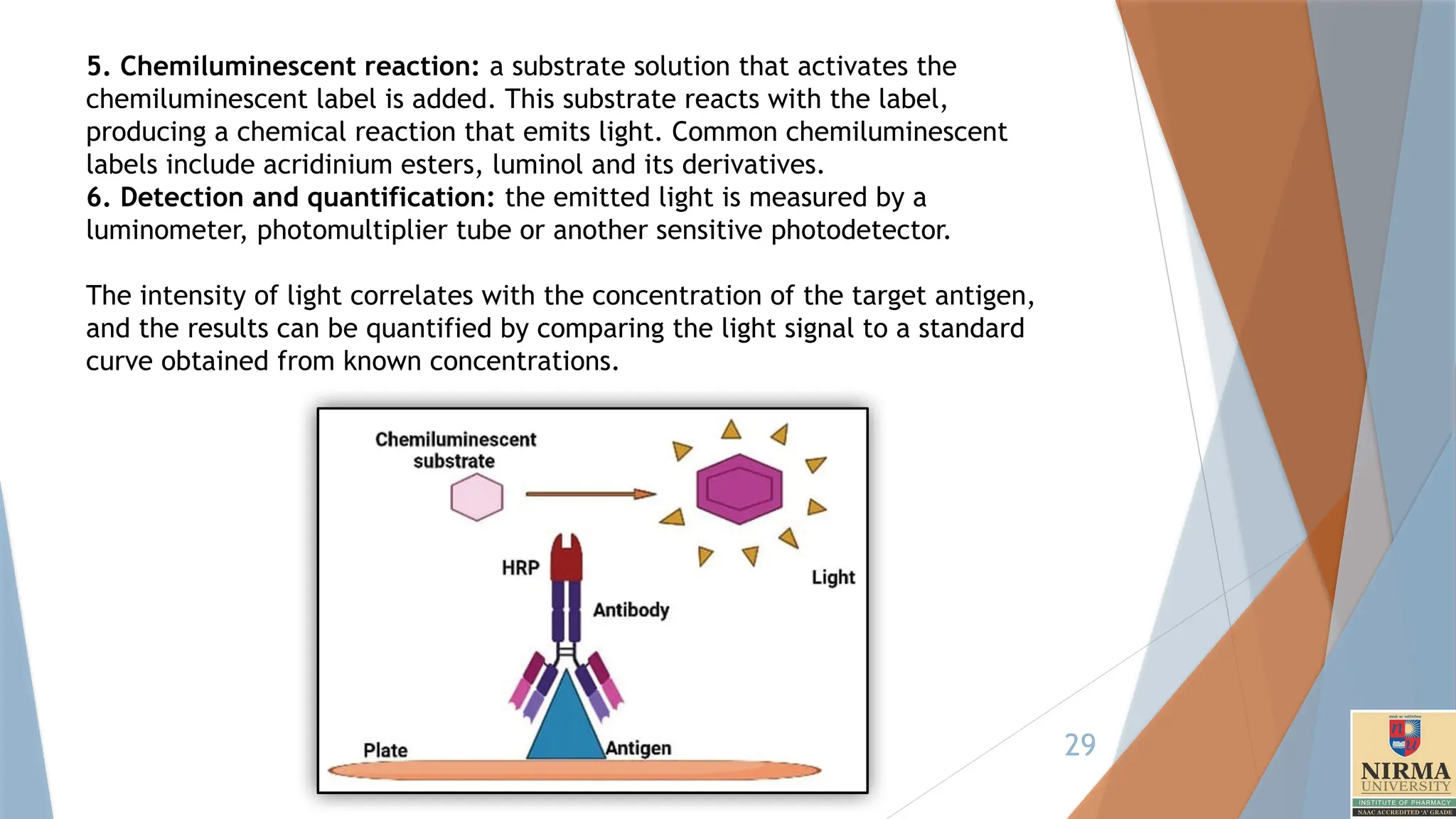
5. Chemiluminescent reaction:a substrate solution that activates the
chemiluminescent label is added. This substrate reacts with the label,
producing a chemical reaction that emits light. Common chemiluminescent
labels include acridinium esters, luminol and its derivatives.
6. Detection and quantification: the emitted light is measured by a
luminometer, photomultiplier tube or another sensitive photodetector.
The intensity of light correlates with the concentration of the target antigen,
and the results can be quantified by comparing the light signal to a standard
curve obtained from known concentrations.
30.
30
1. Plate Coating
Coat a 96-well plate with 100 µL/well of capture antibody diluted in coating buffer (e.g.,
0.1 M carbonate buffer, pH 9.6).
Incubate the plate overnight at 4°C.
Wash the plate 3 times with wash buffer.
Block the wells with 200 µL/well of blocking buffer for 1 hour at room temperature to
reduce non-specific binding.
Wash the plate again 3 times with wash buffer.
2. Sample and Standard Preparation
Prepare gp41 antigen standards in a range of known concentrations using dilution buffer
(PBS + 0.1% BSA).
Add 100 µL/well of standards and samples to the appropriate wells.
Incubate for 1 hour at 37°C.
Wash the plate 5 times with wash buffer.
3. Detection Antibody Binding
Add 100 µL/well of HRP-conjugated anti-gp41 detection antibody diluted in blocking
buffer.
Incubate for 1 hour at room temperature.
Wash the plate 5–7 times with wash buffer to ensure removal of unbound antibodies.
31.
31
4. Chemiluminescent Detection
Add 100 µL/well of Luminol (chemiluminescent substrate).
Incubate for 5–10 minutes at room temperature in the dark.
Measure the luminescence using a plate reader. Record the relative light units (RLU).
the emitted light is measured by a luminometer, photomultiplier tube or another
sensitive photodetector.
The luminescence signal increases proportionally with antigen concentration.
32.
32
Substrates:
1. Luminol andIts Derivatives
Description: Luminol is another widely used substrate that emits light
when oxidized. Its derivatives (such as isoluminol) are also used for
enhanced stability and signal.
Reaction: In the presence of an oxidizing agent like hydrogen peroxide,
and often a catalyst like horseradish peroxidase (HRP), luminol undergoes
an oxidation reaction that produces blue light.
Applications: Luminol is often used with HRP-labelled antibodies,
common in enzyme-linked chemiluminescence immunoassays due to its
high sensitivity.
1. Acridinium Esters
Description: These compounds produce light upon oxidation in an
alkaline environment.
Reaction: In the presence of hydrogen peroxide and an alkaline
environment, acridinium esters undergo oxidation, resulting in the
emission of light.
Applications: Often used in automated immunoassay analysers due to
their high stability and rapid response.
33.
33
5. Immunochromatographic assay
Immunochromatographic assays, often known as lateral flow
assays or rapid tests, are commonly used diagnostic tools for
detecting various biomolecules, such as antibodies, antigens, and
other specific proteins.
They are widely used due to their rapid, simple, and reliable
detection capabilities.
You might have seen these in the form of at-home pregnancy
tests (HCG – Human Chorionic Gonadotropin), COVID-19 tests,
and other point-of-care tests.
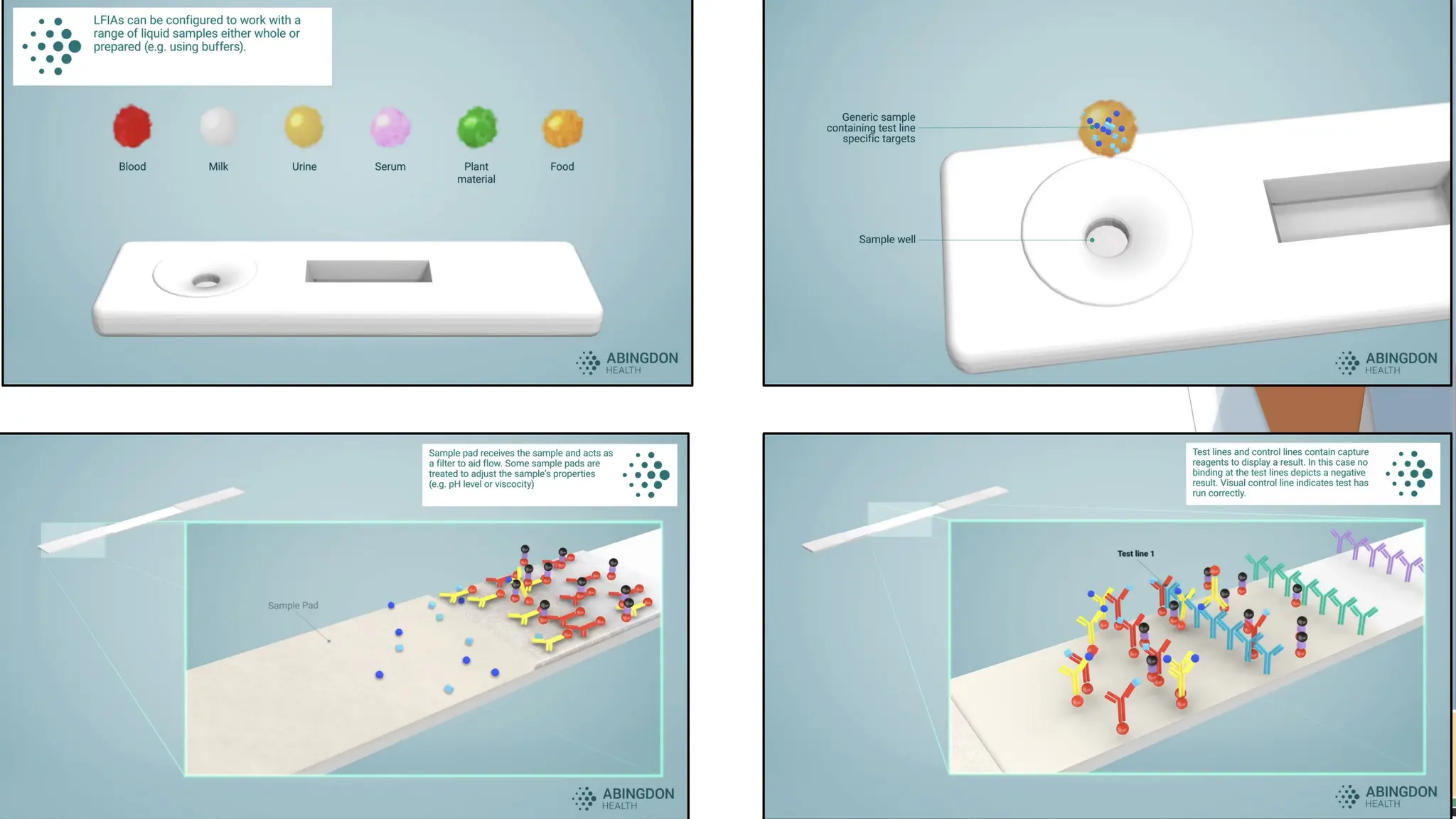
Basic Principle
Immunochromatographic assays work based on the specific
interaction between an antibody and an antigen. These
interactions help capture and visually signal the presence or
absence of the target substance (e.g., a virus or hormone) in the
sample. The test setup uses capillary action to draw a liquid
sample (like blood, urine, or saliva) along a strip of material,
where the sample interacts with embedded antibodies or
antigens.
34.
34
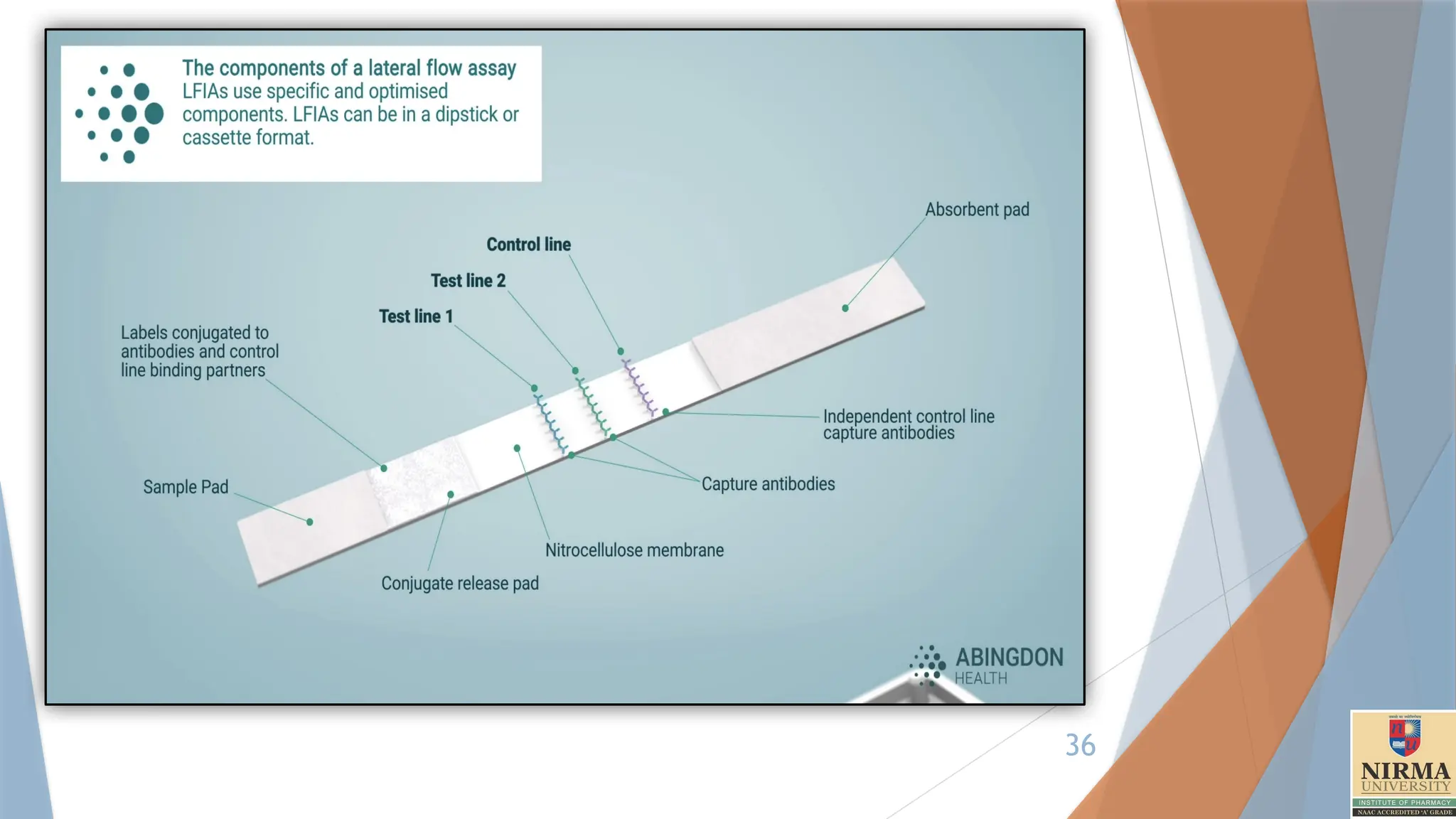
Structure of theTest Strip
A typical immunochromatographic test strip consists of several key
components laid sequentially:
1. Sample Pad: This is where the sample is applied. The pad often
contains substances that help pre-treat or filter the sample to ensure it
flows evenly across the strip.
2. Conjugate Pad: This pad contains antibodies (or antigens, depending
on the target) conjugated to coloured particles, such as gold
nanoparticles or latex microspheres. These particles are responsible for
the visual signal when the target is detected. The antibodies in the
conjugate pad are specific to the target molecule, and they are designed
to bind with it if present.
35.
35
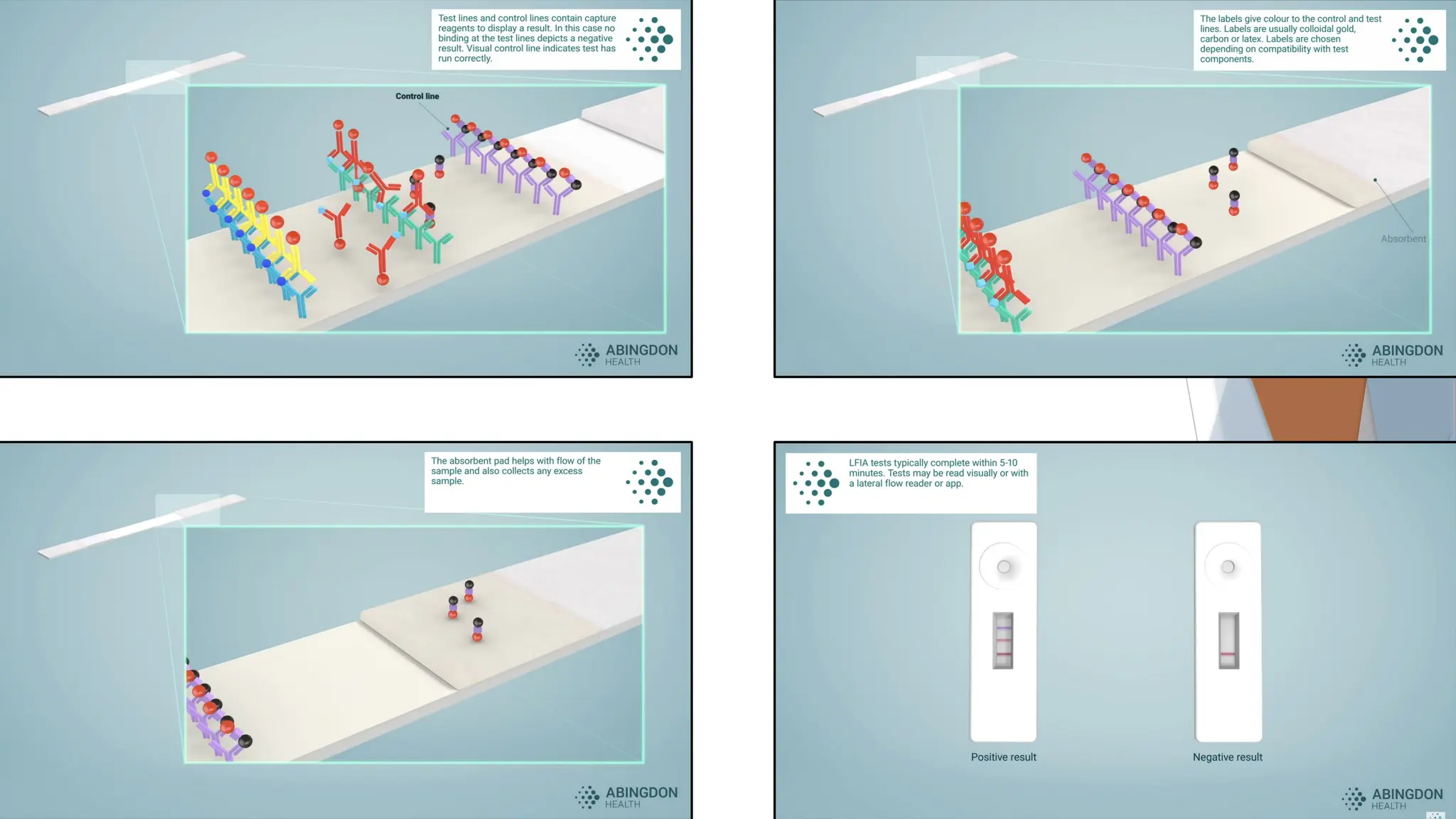
3. Nitrocellulose Membrane:This is the main area of the strip, where
detection occurs. The membrane typically has:
• Test Line: This contains immobilized antibodies that are specific to
the target antigen or antibody. When the target analyte binds to the
conjugated antibodies from the conjugate pad, it will also bind here,
creating a visible line if the target is present.
• Control Line: This line contains antibodies that react with
components in the conjugate pad regardless of whether the target
is present. This line appears in every test to confirm that the sample
has migrated correctly and that the test is functioning.
4. Absorbent Pad: This pad at the end of the strip helps absorb excess
liquid, pulling the sample through the strip by capillary action and ensuring
proper flow.
39
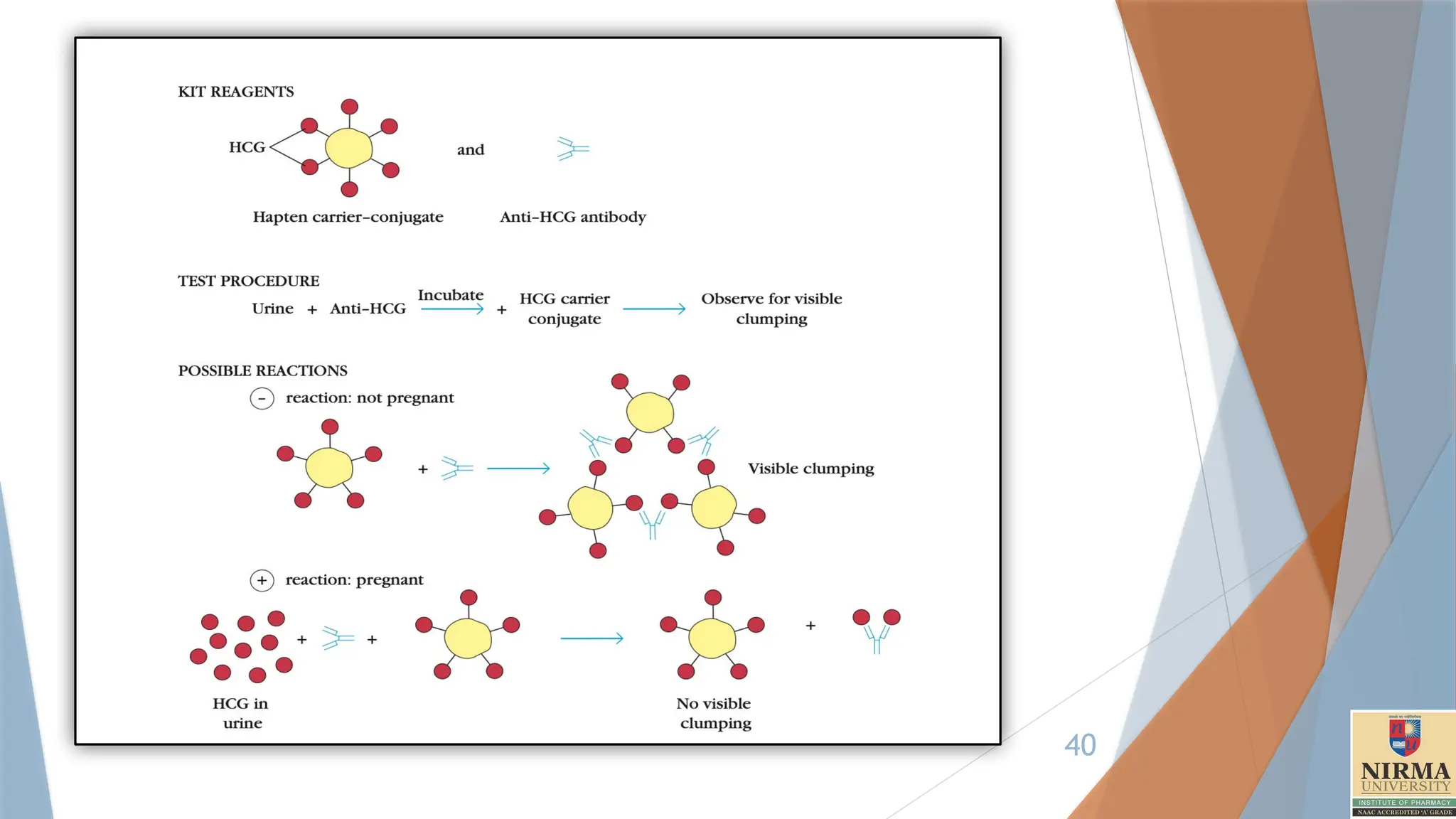
A modificationof the agglutination reaction, called agglutination inhibition, provides
a highly sensitive assay for small quantities of an antigen.
For example, one of the early types of home pregnancy test kits included latex
particles coated with human chorionic gonadotropin (HCG) and antibody to HCG. The
addition of urine from a pregnant woman, which contained HCG, inhibited
agglutination of the latex particles when the anti-HCG antibody was added; thus the
absence of agglutination indicated pregnancy.
Agglutination inhibition assays can also be used to determine whether an individual is
using certain types of illegal drugs, such as cocaine or heroin. A urine or blood
sample is first incubated with antibody specific for the suspected drug. Then red blood
cells (or other particles) coated with the drug are added. If the red blood cells are not
agglutinated by the antibody, it indicates the sample contained an antigen recognized
by the antibody, suggesting that the individual was using the illicit drug.
One problem with these tests is that some legal drugs have chemical structures
similar to those of illicit drugs, and these legal drugs may cross-react with the
antibody, giving a false-positive reaction. For this reason a positive reaction must be
confirmed by a nonimmunologic method.
41
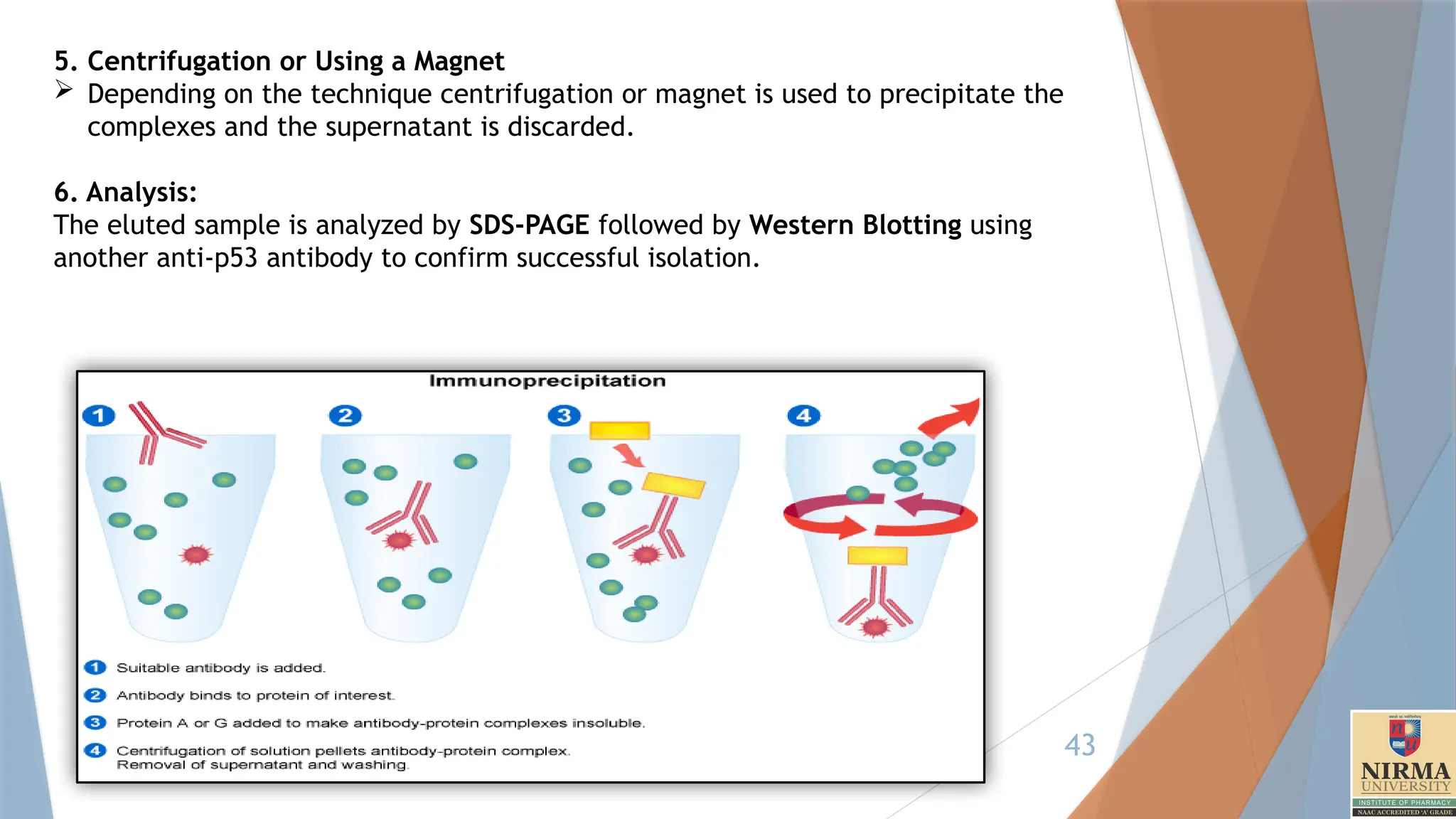
6. Immunoprecipitation
Immunoprecipitation(IP) is a widely used technique in biochemistry and
molecular biology that allows for the isolation and enrichment of a specific
antigen (protein) from a complex mixture, like a cell lysate, by using an
antibody that specifically binds to that antigen.
Principle:
The principle involves the formation of an antigen-antibody complex, where
an antibody binds to its target protein. This complex is then “precipitated”
(pulled out of solution) by adding a secondary reagent like protein A or G
beads, which bind to the antibody and get precipitated on centrifugation.
Another special version involves precipitation through the use of magnetic
beads. After washing away unbound components, the protein of interest
can be analysed further, often by Western blotting or mass spectrometry.
Example: Immunoprecipitation of p53 protein(Tumour suppressor protein).
Here prewashed Protein G beads are used.
42.
42
Procedure:
1. Preparation ofcell lysate
To release proteins from a cell, they are lysed using a lysis buffer such as NP-40
or Triton X-100 to solubilize the membrane.
Protease inhibitors and phosphatase inhibitors are used to prevent degradation
of proteins. (Leupeptin and Aprotinin) (Sodium fluoride (NaF))
In ChIP technique, Formaldehyde is used to covalently bind DNA and proteins to
identify DNA sequences associated with specific proteins (Transcription Factors).
2. Antibody Incubation:
A monoclonal anti-p53 antibody is added to the lysate and incubated overnight at
4°C with gentle mixing.
3, Bead binding:
Prewashed Protein G agarose beads are added to the mixture and incubated for 1-2
hours at 4°C to capture the antibody – p53 complex.
4. Washing the beads
Carefully wash the beads several times (usually 3-5 times) with cold lysis buffer or
a low-salt wash buffer. This helps remove non-specifically bound proteins.
43.
43
5. Centrifugation orUsing a Magnet
Depending on the technique centrifugation or magnet is used to precipitate the
complexes and the supernatant is discarded.
6. Analysis:
The eluted sample is analyzed by SDS-PAGE followed by Western Blotting using
another anti-p53 antibody to confirm successful isolation.
44.
44
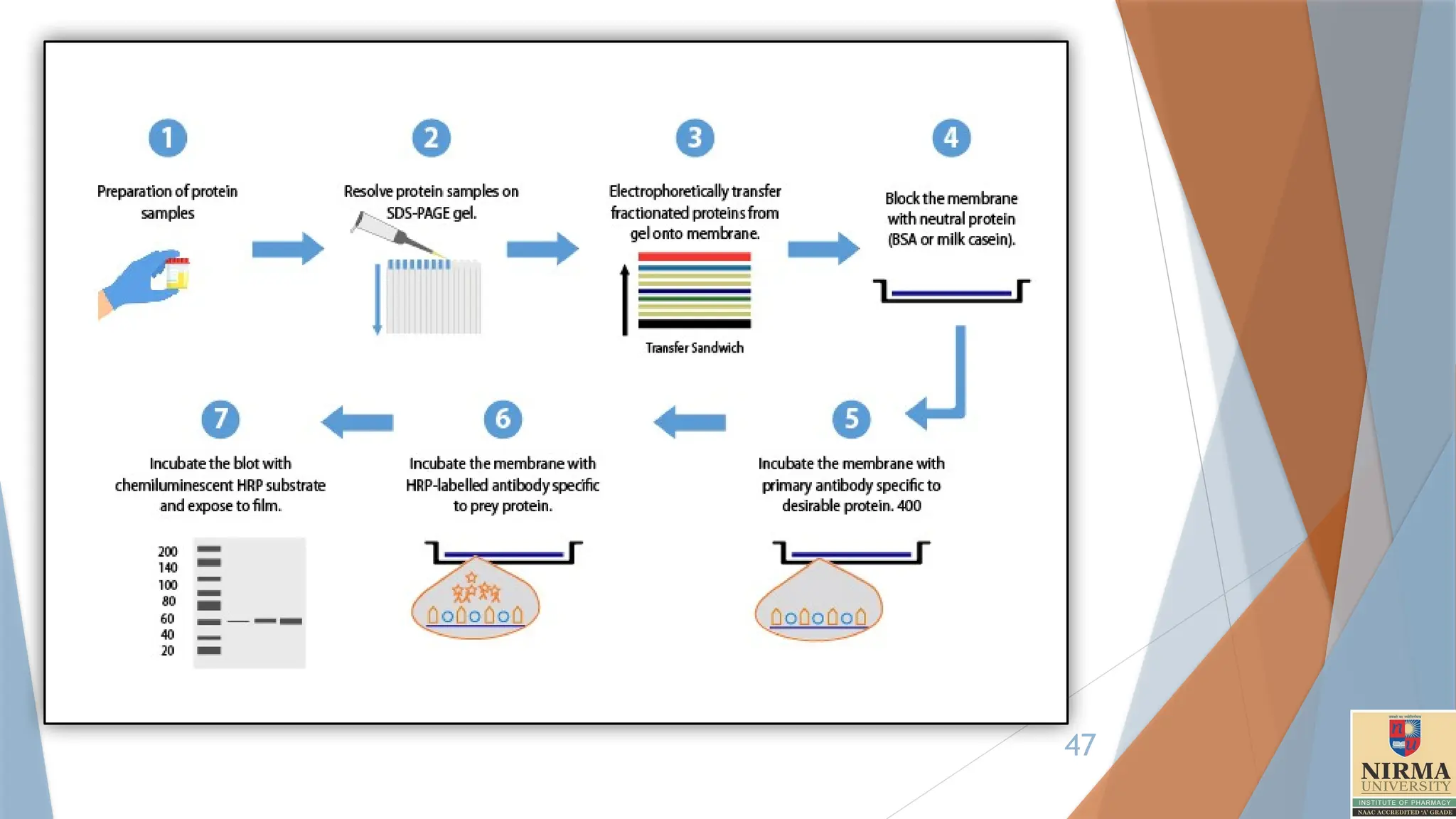
7. Western blotting
Thewestern blot (sometimes called the protein immunoblot), or western
blotting, is a widely used analytical technique in molecular
biology and immunogenetics to detect specific proteins in a sample of tissue
homogenate or extract.
1. Cell lysis
Cells or tissues are lysed to release proteins, typically using a lysis buffer
containing detergents, salts, and protease inhibitors to prevent protein
degradation.
2. Gel Electrophoresis
Proteins are separated by size using sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE). SDS is a detergent that denatures proteins
and gives them a uniform negative charge.
Loading and Running the Gel: Protein samples are loaded into wells in the gel
along with a molecular weight marker or ladder for size reference. An electric
current is applied, causing the proteins to migrate through the gel toward the
positive electrode.
45.
45
Separation: Theproteins separate based on size, with smaller proteins moving
faster and larger proteins moving slower through the gel matrix.
3. Protein Transfer
After electrophoresis, the separated proteins are transferred from the gel onto
a membrane, typically made of nitrocellulose or polyvinylidene fluoride
(PVDF).
This step is done using a wet transfer method.
Wet Transfer: The gel and membrane are sandwiched between filter papers
soaked in transfer buffer, and an electric current is applied to transfer proteins.
Transfer buffer consists of Tris base (pH-8.3), Glycine and 20% Methanol
which is responsible for removing the attached SDS molecules on the proteins
allowing it to bind properly.
This step creates an exact replica of the protein pattern on the membrane,
making it accessible for antibody binding in the next steps.
4. Blocking
The membrane is incubated in a blocking buffer (typically containing non-fat
milk or BSA in buffer) to prevent non-specific binding of antibodies to the
membrane.
Blocking ensures that the antibodies will bind only to specific protein sites and
not to other parts of the membrane, reducing background noise in the final
detection.
46.
46
5. Primary AntibodyIncubation
The membrane is incubated with a primary antibody that specifically binds to the
target protein.
6. Washing
The membrane is washed several times to remove unbound primary antibody.
Washing is typically done with a buffer containing a mild detergent (like Tween-20) to
reduce non-specific interactions.
7. Secondary Antibody Incubation
The membrane is then incubated with a secondary antibody that recognizes and
binds to the primary antibody.
This secondary antibody is often conjugated to an enzyme, such as horseradish
peroxidase (HRP) or alkaline phosphatase (AP), which allows for signal detection.
The use of a secondary antibody amplifies the signal, as multiple secondary
antibodies can bind o each primary antibody, improving sensitivity.
Again washing is performed to remove the unbound secondary antibody.
9. Detection
The membrane is treated with a substrate that reacts with the enzyme
linked to the secondary antibody, producing a detectable signal.
Common Detection Methods:
Chemiluminescence, Chromogenic detection, Fluorescent detection
48
A familiar andwidely used example in Western blotting is the detection of GAPDH
(Glyceraldehyde-3-phosphate dehydrogenase), another housekeeping protein often
used as a loading control. GAPDH is typically found at approximately 37 kDa in
mammalian cells. Here's the same procedure applied to detecting GAPDH:
1. Sample Preparation
Lysis:
• Collect cells (e.g., HeLa cells) and lyse them in RIPA buffer containing protease
inhibitors to prevent protein degradation.
• Centrifuge at 12,000 × g for 10–15 minutes to remove debris and collect the
supernatant containing proteins.
Quantification:
• Perform a protein quantification assay (e.g., BCA assay) and adjust all samples
to the same concentration (e.g., 2 mg/mL).
49.
49
2. SDS-PAGE
GelPreparation:
• Use a resolving gel with 10% acrylamide, ideal for separating proteins in the 20–
60 kDa range.
• Load 20 µg of protein per well after mixing with 4× Laemmli buffer and boiling at
95°C for 5 minutes.
Electrophoresis:
• Run the gel at 80 V (stacking gel) and then increase to 120 V for the resolving
gel. The GAPDH band will migrate close to the 37 kDa marker in the protein
ladder.
3. Protein Transfer
Transfer Setup:
• Transfer proteins to a PVDF membrane pre-activated in methanol using a wet
transfer system at 100 V for 1 hour.
Confirm Transfer:
• Stain the membrane with Ponceau S to visualize transferred proteins, ensuring
successful protein transfer.
4. Blocking
Incubate the membrane in 5% non-fat milk dissolved in TBS-T (Tris-buffered saline
with Tween-20) for 1 hour at room temperature.
50.
50
5. Primary AntibodyIncubation
Incubate with a monoclonal anti-GAPDH antibody (e.g., mouse-derived, diluted
1:1000 in TBS-T containing 1% BSA) overnight at 4°C.
6. Washing
Wash the membrane 3× with TBS-T for 5–10 minutes each time to remove unbound
primary antibody.
7. Secondary Antibody Incubation
Incubate with an HRP-conjugated anti-mouse secondary antibody (1:5000 dilution in
TBS-T) for 1 hour at room temperature.
8. Washing
Repeat the washing step to remove excess secondary antibody.
9. Detection
Chemiluminescence:
• Incubate with ECL substrate for HRP and expose the membrane to a
chemiluminescent imager or X-ray film.
Visualize GAPDH:
• GAPDH appears as a clear band at ~37 kDa on the developed image.
51.
51
Why GAPDH?
GAPDHis a cytosolic protein with stable expression in most cells under normal
conditions, making it ideal for verifying equal sample loading.
This example is especially relevant in studies involving protein expression changes in
diseases like cancer or neurodegenerative conditions.
DNA repair: GAPDH participates in DNA replication and repair
Apoptosis: GAPDH promotes cellular apoptosis under certain conditions
Gene expression: GAPDH is involved in gene expression and transcriptional
modulation
Cytoskeletal organization: GAPDH modulates the organization and assembly of the
cytoskeleton
Inflammation: GAPDH is involved in innate immunity and inflammatory processes
Neurodegenerative disease: GAPDH has been implicated in neurodegenerative
disease
Prostate cancer: GAPDH has been implicated in prostate cancer








![17
The plate is incubated (8-10 Hours) to allow the antibody to adhere to
the surface of the wells.
Excess antibody is washed away, and unbound sites on the plate are
blocked using a blocking agent (e.g., bovine serum albumin-BSA or Skim
milk) to prevent nonspecific binding.
2. Sample Addition:
The patient's blood or plasma sample is added to the wells. If the HIV
antigen is present, it will bind to the immobilized capture antibody.
The plate is incubated to allow sufficient binding time (1-2 hours at room
temp.), and unbound material is washed away.
3. Addition of Detection Antibody:
A secondary antibody, labelled with an enzyme (e.g., horseradish
peroxidase [HRP]), is added to the wells. This detection antibody binds to
a different epitope of the HIV antigen, completing the "sandwich."
Unbound detection antibody is washed away after incubation.](https://image.slidesharecdn.com/9-250404122538-c1993e32/75/Protocols-for-different-types-of-Immunoassay-17-2048.jpg)
![18
4. Enzyme-Substrate Reaction:
A substrate for the enzyme (e.g., TMB [3,3’,5,5'-tetramethy|benzidine]) is
added. The enzyme catalyses a reaction that produces a colour change.
The intensity of the colour is directly proportional to the amount of HIV
antigen in the sample.
5. Measurement:
The colour intensity is measured using a spectrophotometer at a specific
wavelength (e.g., 450 nm).
A standard curve is used to quantify the antigen concentration, if needed.
Applications of HIV/AIDS Diagnosis:
Sandwich ELISA is widely used in combination with other tests ( e.g.
antibody detection ELISA, Western Blot, or PCR) to confirm HIV status. It
plays a crucial role in early detection, monitoring disease progression, and
evaluating the effectiveness of Antiretroviral therapy (ART).](https://image.slidesharecdn.com/9-250404122538-c1993e32/75/Protocols-for-different-types-of-Immunoassay-18-2048.jpg)