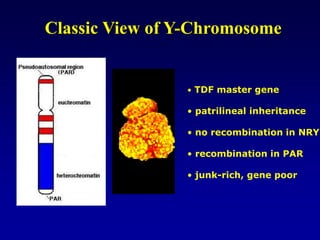
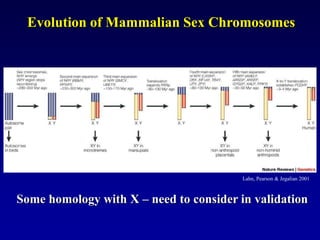
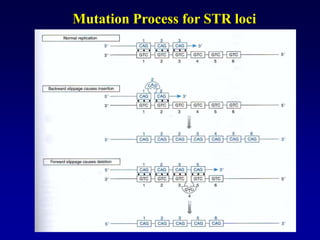
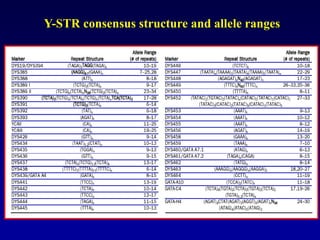
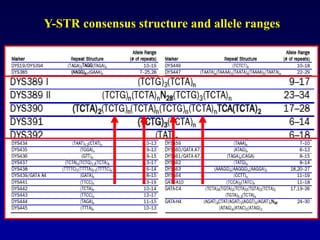
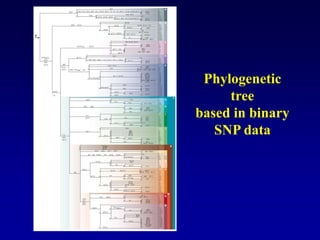
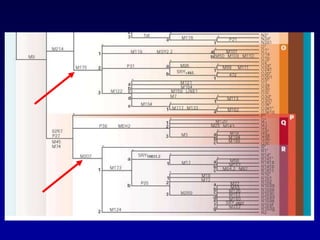
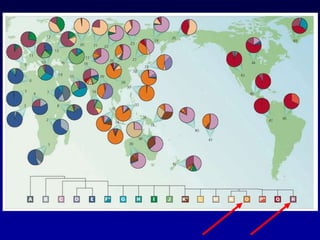
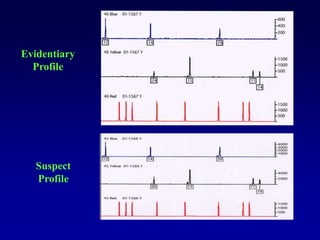
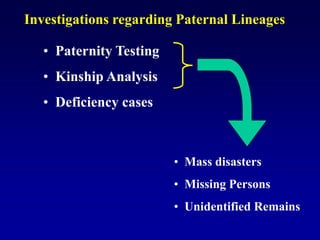
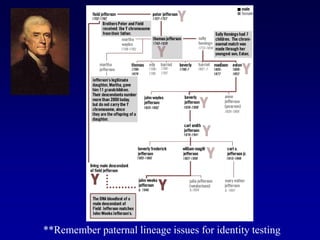
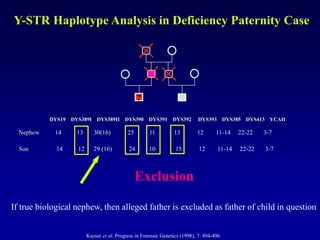
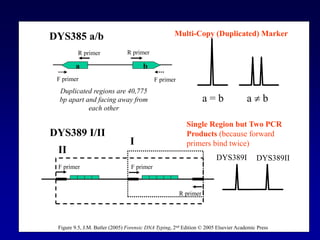
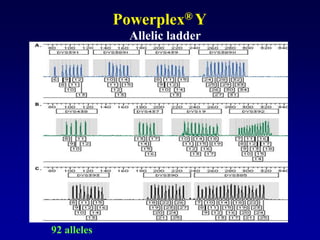
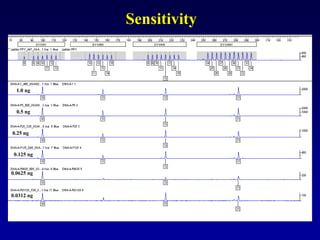
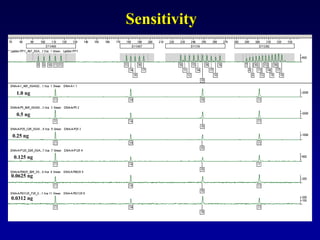
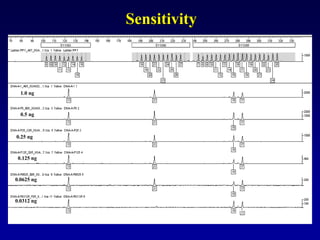
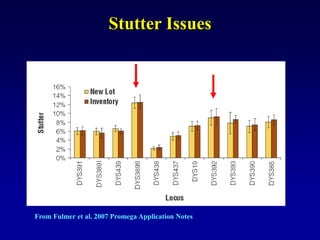
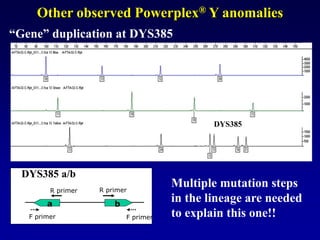
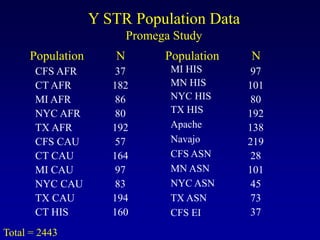
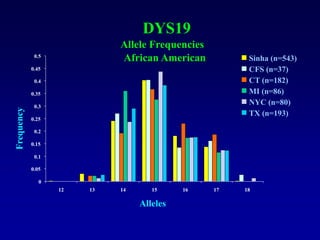
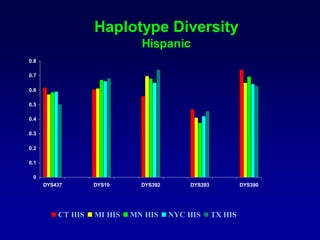
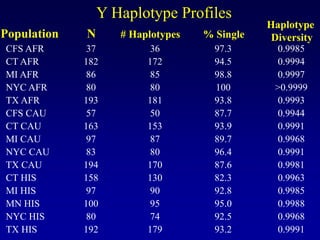
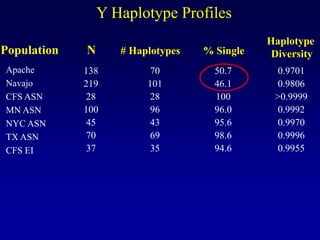
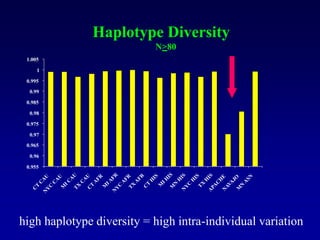
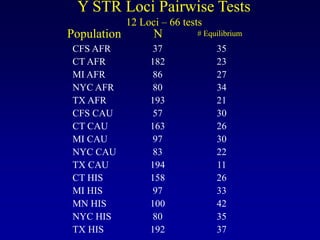
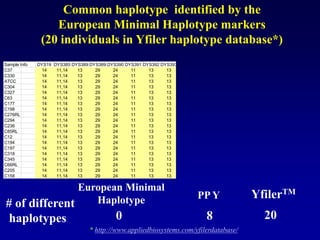
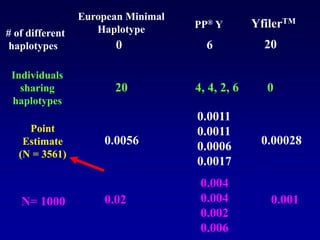
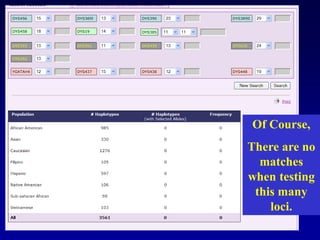
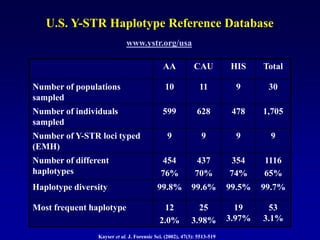
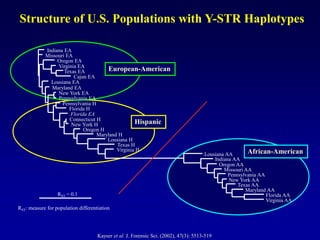
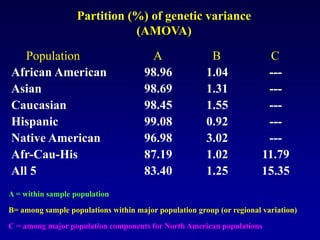
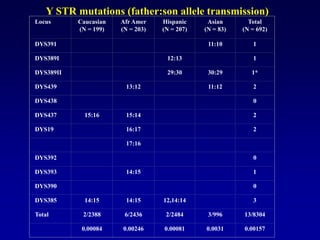
This document discusses applications of forensic genetics using Y-STR DNA analysis. It begins by describing characteristics of the human Y chromosome, including its size, gene content, and lack of recombination. It then discusses Y-STR polymorphisms and markers used in forensic kits. Examples are given of cases where Y-STR analysis was used, such as analyzing mixed male-female samples or determining paternity. Guidelines for interpretation and population studies using the PowerPlex Y kit are discussed. Common issues like sensitivity, mixtures, and anomalies are also summarized.
















































































































![LR =
1
2[Pr(H1)Pr(H8) + Pr(H2)Pr(H7) + Pr(H3)Pr(H6) + Pr(H4)Pr(H5)]
Mixture
Likelihood Ratio](https://image.slidesharecdn.com/yworkshopwiplanz3-240222183556-4849c10c/85/Y_Workshop_WI_planz-3-ppt12345789999987543-152-320.jpg)










































































