Protein Structure, Ramachandran Plot, alpha, beta helix,
1.
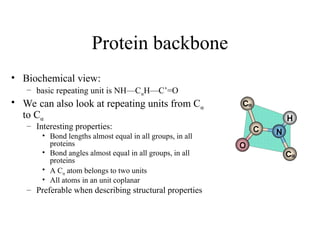
Protein backbone
• Biochemicalview:
– basic repeating unit is NH—CαH—C’=O
• We can also look at repeating units from Cα
to Cα
– Interesting properties:
• Bond lengths almost equal in all groups, in all
proteins
• Bond angles almost equal in all groups, in all
proteins
• A Cα atom belongs to two units
• All atoms in an unit coplanar
– Preferable when describing structural properties
2.
Protein backbone
• Geometric/Structuralview: polypeptide chain divided into
– Peptide units
• Cα atom and carboxyl group of residue i
• Amino group and Cα atom of residue i+1
• Are rigid groups
• Rotation on bond C-N is prevented by energy barrier
• Peptide units are joined by covalent bonds between Cα atoms. Thus
– Peptides can rotate along 2 bonds:
• N-Cα and Cα-C
– Two dihedral angles for each unit: Ф (Phi) and Ψ (Psi)
• Two degrees of freedom per unit
• Determine the conformation of the backbone
3.
Dihedral angles andregular
structures
• Repeating values of Ф and Ψ along the main chain result in regular structure
– repeating values of Ф =-57o
and Ψ =-47o
give a right-handed helical fold (α-helix)
– repetitive values of Ф[-110,-140] and Ψ[+110,+135] give sub chains with
conformations that allow interactions between nearby parallel segments (β-sheet)
• Most combinations of Ф and Ψ angles are not allowed
– Allowed conformations plotted as 2-D chart
• Ramachandran plot
4.
Secondary Structure
• definedby patterns of hydrogen bonds between backbone amide groups
– sidechain-mainchain and sidechain-sidechain hydrogen bonds are irrelevant
• The amino acids in the interior/core of a globular protein have hydrophobic
side chains
– Water soluble proteins fold to pack hydrophobic side chain into interior
– Results in hydrophobic core and hydrophilic surface
– The main chain must fold into interior, too
• Main chain is hydrophilic:
– N--H: hydrogen bond donor
– C=O: hydrogen bond acceptor
– These groups must be neutralized by formation of H bonds secondary structure
• Secondary structure
– α-helices
– β-sheets
– form rigid and stable frameworks
5.
α-Helix
• Righthanded coiledconformation
– backbone N-H group i+4 forms hydrogen bonding with
backbone C = O group i
– 3.6 residues per turn (5.4 Å, 1.5 Å per residue)
• Variations, with chain more loosely or tightly coiled are
possible (i+3 or i+5 instead of i+4) but not often
– backbone (φ, ψ) dihedral angles around (-60o
,-45o
)
– Sum of φ and ψ angles of consecutive residues about
105o
• Has between 4 to 40 residues
• All H bonds point in the same direction
– Aligned along helical axis
– Dipole moments for residues are aligned along axis
• Net dipole for α-helix (+ at N-H end and – at C=O end)
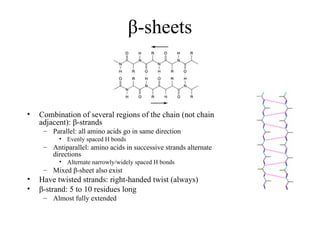
β-sheets
• Combination ofseveral regions of the chain (not chain
adjacent): β-strands
– Parallel: all amino acids go in same direction
• Evenly spaced H bonds
– Antiparallel: amino acids in successive strands alternate
directions
• Alternate narrowly/widely spaced H bonds
– Mixed β-sheet also exist
• Have twisted strands: right-handed twist (always)
• β-strand: 5 to 10 residues long
– Almost fully extended
From secondary structureto structure
• Protein structure: built from secondary
structures
– Connected by loop regions
• Various lengths
• Irregular shape
• Are at the surface of the protein
• Reach in charged and polar residues
– Easier to predict!
• In homologous proteins almost always insertions and
deletions occur in the loop regions.
10.
Structure Motifs
• Secondarystructures connected to form
motifs
– α-helices and β-sheets in a motif
• Adjacent in the 3-dimensional structure
• Connected bu loop regions
• Combinations of motifs and secondary
structures domains
11.
• Tertiary structure:
–Arrangement of secondary structure
– Structural domains
• Quaternary structure
– More than one polypeptide folded together
• Native conformation: direct consequence of
– primary structure
– chemical environment
• water based
• oily interior of a cell membrane
– So far, no reliable computational method exists to predict
the native structure from the amino acid sequence
12.
Structure Classes
• Proteinstructure four classes:
– α-domains
• core built up only from α-helices
– β-domains
• core built up only from (usually 2) antiparallel β-sheets
– α/β-domains
• mostly β-α-β motifs
– (mostly) parallel β-sheets surrounded by α-helices
– α+β-domains (few cases)
• antiparallel β-sheet packed against α-helices

![Dihedral angles and regular
structures
• Repeating values of Ф and Ψ along the main chain result in regular structure
– repeating values of Ф =-57o
and Ψ =-47o
give a right-handed helical fold (α-helix)
– repetitive values of Ф[-110,-140] and Ψ[+110,+135] give sub chains with
conformations that allow interactions between nearby parallel segments (β-sheet)
• Most combinations of Ф and Ψ angles are not allowed
– Allowed conformations plotted as 2-D chart
• Ramachandran plot](https://image.slidesharecdn.com/proteinstructure-251005154507-a32ee800/85/Protein-Structure-Ramachandran-Plot-alpha-beta-helix-3-320.jpg)