
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Protein Purification Lecture
Similar to Protein Purification Lecture (20)
More from ouopened
More from ouopened (17)
Recently uploaded
Recently uploaded (20)
Protein Purification Lecture
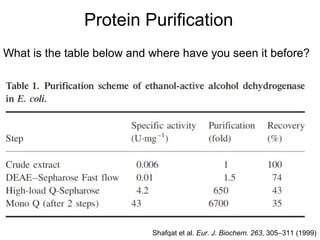
- 1. Protein Purification What is the table below and where have you seen it before? Shafqat et al. Eur. J. Biochem. 263, 305-311 (1999)
- 2. Overall Strategy Molecular characteristics of target protein Look for the properties that distinguish the target from other proteins Target features charge/hydrophobicity solubility & stabilization size Define a way of identifying the presence of your protein, and of assessing its purity, before you start purification
- 3. Protein Purification I. Source II. Method of lysing the cells III. Protecting the protein from degradation IV. Means to keep track of your protein – activity assay, protein purification table V. Early Stages – gentle fractionation steps to handle largish volumes. VI. Middle Stages – chromatographic separation(s) VII. Final (Polishing) Stages Reading: N & B Chapters 6 & 7
- 4. I. Source What source will you use to obtain the protein of interest? A) Natural – some plant or animal tissue B) Recombinant – a gene for the protein of interest that has been cloned into a vector and transformed into expression cells
- 5. Some other considerations Structural Integrity “native” versus “non-native” conformation denaturation/renaturation protein folding/re-folding subunit dissociation/re-association
- 6. Choice of Buffer The pKa of the buffer should be within 0.5 unit of the desired pH Potential interactions with a column matrix of any resins you might use Avoid UV-absorbing buffers if you plan to use a UV detector The ionic strength and salt composition must be chosen according to the stability of the protein and the detergent
- 7. Typical Solution Components Buffer Salt Detergents (sometimes) Glycerol, sugars Metal chelators Protease inhibitors (usually) Reducing agents (often) Ligands (sometimes)
- 8. Centrifuge Be very careful using the centrifuge. Samples must be balanced. Clean up any spills, especially those that have ammonium sulfate, which is very corrosive.
- 9. On December 16, 1998, milk samples were running in a Beckman L2-65B ultracentrifuge using a large aluminum rotor (a rotor is a large metal object that holds the individual sample tubes and is connected to the spin drive of the centrifuge). The rotor had been used for this procedure many times before. Approximately one hour into the operation, the rotor failed due to excessive mechanical stress caused by the "G" forces of the high rotation speed. The subsequent explosion completely destroyed the centrifuge (Images 1 & 2). The safety shielding in the unit did not contain all the metal fragments. The half-inch thick sliding steel door on top of the unit buckled allowing fragments, including the steel rotor top, to escape (Image 3). Fragments ruined a nearby refrigerator and an ultra-cold freezer in addition to making holes in the walls and ceiling. The unit itself was propelled sideways and damaged cabinets and shelving that contained over a hundred containers of chemicals (Image 4). Fortunately, sliding cabinet doors prevented the containers from falling to the floor and breaking. A shock wave from the accident shattered all four windows in the room. The shock wave also destroyed the control system for an incubator and shook an interior wall causing shelving on the wall to collapse (Image 5). Fortunately the room was not occupied at the time and there were no personal injuries.
- 10. We will have more to say about centrifugation, especially analytical ultracentrifugation, during a later lecture
- 11. II. Method of lysing the cells How will you “break open” the cells of the tissue or the bacterial cells? Not a trivial matter. A) Mechanical disruption – homogenizing the tissue with a Waring blendor (note the spelling). B) French Press C) Sonication D) Freeze Thaw Cycles E) Enzymatic – e.g., Lysozyme or Chemical Methods
- 12. Frederick Waring and his “blendor” http://extranet.libraries.psu.edu/psul/waring/blendor.html
- 13. A French Press, but not the one we are interested in http://upload.wikimedia.org/wikipedia/commons/9/91/French_press.jpg
- 14. This is the French Press we are interested in Analytical Biochemistry 321 (2003) 276–277
- 15. Some examples of sonicators http://en.wikipedia.org/wiki/Sonication http://www.fishersci.com/wps/portal/PRODUCTDETAIL?aid=2819374
- 16. III. Protecting the protein from degradation OK, so you have figure out a way or method to break open the cells, now how are you going to protect your protein from degradation? A) Add PMSF (phenylmethylsulfonylfluoride) to inhibit proteases B) Add other protease inhibitors C) High or low pH to inhibit proteases, working quickly D) Use “Expression” cells
- 17. IV. Means to keep track of your protein – activity assay, protein purification table How are you going to know where your protein is? Is it in the pellet or the supernatant following a centrifugation step? A) Activity assay – a beacon in the “soup”. Activity versus Specific Activity. B) At each step you save a sample and later construct a protein purification table. C) Use of PAGE – subject for later, but not ideal as it is too time consuming.
- 18. A410 N&B Fig. 7-1 Activity Assay
- 19. "Specific Activity" Definition: Units of enzyme activity per mg protein 1 Unit = amount of enzyme that will convert one μmole of substrate to product in one minute at a given pH (optimum value) and temperature (usually 25°C or 37°C). Specific activity is used as an estimate of enzyme purity.
- 20. Example You have total of 5 mL of 2 mg/mL protein. From a 3 mL assay containing 1.5 mL of 50 mM substrate and 100 mL of your protein you obtain ΔA/Δt = 0.1 min-1. Calculate the specific activity of the enzyme in the sample. Assume that the substrate has the following extinction coefficient: e410 nm = 18.8 mM-1 cm-1. Report your answer to one significant figure.
- 21. Activity vs. Specific Activity
- 22. Purification Table (example) (see N&B p. 172 for more details)
- 23. V. Early Stages – gentle fractionation steps to handle largish volumes. How can I gently separate my protein from a number of other contaminants using relatively gentle methods? A) Salting in/Salting out – the use of ammonium sulfate to gently fractionate your protein B) How to “desalt” your protein following ammonium sulfate precipitation? - Use of gel-filtration in the course mode - Use of dialysis
- 24. Perturbations of the solvent-protein interactions Major forces within a polypeptide chain and between chains that drive a protein to a stable conformation -Ion-ion -Ion-dipole (hydrogen bonds) -Hydrophobic Perturbations of the solvent-protein interactions can cause transitions by disrupting the “old” interactions and promoting formation of new ones.
- 25. Perturbations that cause transitions A rise in temperature (can weaken the strength of dipolar interactions and favor formation of hydrophobic interactions) pH-dependent protonation and deprotonation Large amount of a salt or organic solvents
- 26. Precipitation of proteins by salts Precipitation occurs by: -neutralization of surface charges by the salt -reducing the chemical activity of the protein -diminishing the effective concentration of water This is called “salting out” of proteins
- 27. Principle behind salting out of proteins 1. The concentration of any salt necessary to cause precipitation of a particular protein is related to the number and distribution of charges and of hydrophobic residues exposed and rendered dominant as the charges are neutralized. 2. This property is exploited to separate some proteins from others by precipitation at high salt concentrations. 3. As salt concentration increases, protein solubility decreases. 4. Net effect is dehydration of proteins which promotes self-association and aggregation
- 28. Ammonium sulfate precipitation ©Wilbur H. Campbell, 1996
- 29. Ammonium Sulfate Fractionation Why use ammonium sulfate (NH4)2SO4 as opposed to other salts? • well tolerated by most proteins • highly soluble (up to ~ 4 M) • does alter pH significantly • relatively inexpensive
- 32. VI. Middle Stages – chromatographic separation(s) What chemical properties of your protein can you take advantage of? What is the pI of your protein? A) Ion-Exchange Chromatography -anion exchange -cation exchange B) Affinity Chromatography -resin contains ligand or substrate covalently attached -use of an affinity tag (e.g., “His-tag”)
- 33. Affinity Chromatography The basic idea is to use a resin that contains a molecular fragment that interacts specifically with the species of interest (or a relatively small class of compounds that have similar chemical properties). The species of interest binds to the affinity resin while the other compounds present in the mixture pass through the column. The species of interest can then be eluted.
- 35. Affinity Chromatography: another view
- 36. Affinity Chromatography Continued: Purification of His-tagged proteins -from “The QIAexpressionistTM”, by Qiagen, 2003
- 37. Two different types of nickel affinity resins: NTA – nitrilotriacetic acid IDA – iminodiacetic acid -from “The QIAexpressionistTM”, by Qiagen, 2003
- 38. Binding of the His-tagged protein to the Ni-NTA resin. -from “The QIAexpressionistTM”, by Qiagen, 2003
- 39. VII. Final (Polishing) Stages Are there still some nagging contaminants present? A) Rerunning an ion-exchange column with a slightly different matrix but same functional group B) Gel-filtration chromatography – high resolution mode C) Ultra-filtration/centrifugal concentrators
- 40. Ultrafiltration Use centrifugation to achieve solvent flux through the membrane Applications: -Desalting and buffer exchange -Concentrating and purifying proteins, antibodies and nucleic acids (alternative to EtOH precipitation)
- 41. Centrifugal Concentrators Centrifugation forces buffer through the membrane, retaining molecules larger than the cut-off within the sample tube. Repeated dilution and concentration can achieve rapid buffer exchange.
- 42. Fundamentals of Ligand Binding: Ligands collide with their targets, at a rate of kon. Usually this rate is diffusion limited and occurs about a rate of 108 – 109 M-1 s-1. The ligand leaves its binding site at a rate that depends on the strength of interaction between the ligand and the binding site. Off-rates (koff) range from 106 s-1 (weak binding) to 10-2 s-1 (strong binding). The equilibrium constant for binding is given by: or
- 43. Define the fractional saturation (Y) as follows: But, since
- 44. Y
- 45. The binding constant of a protein for a small ligand may be determined by equilibrium dialysis