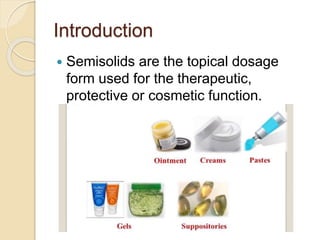
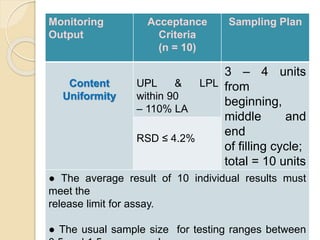
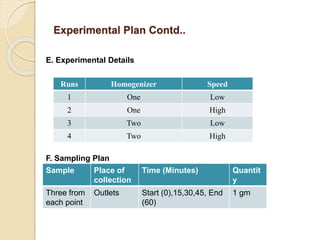
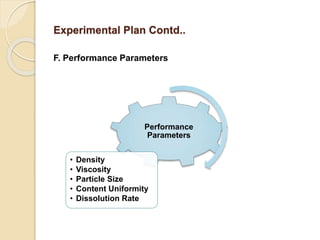
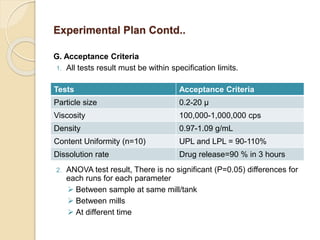
This document outlines the process validation approach for manufacturing semi-solid dosage forms. It discusses pre-requisites like equipment and facility qualification. Critical process parameters identified include mixing speed, homogenization time, and storage conditions. The document proposes a prospective validation approach using 3 consecutive successful batches. It recommends sampling plans and acceptance criteria for validation including testing bulk and finished products for properties like viscosity, particle size, and content uniformity. An experimental plan is provided to validate the homogenization step focusing on achieving homogeneous bulk mixtures.





















































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)

